
第六章分子的结构与性质 6-1键参数 61-1键能 1.键能(E)一一衡量原子之间形成的化学键强度(键牢固程度)的键参数。粗略而 言,是指在标准状态下气态分子每断裂1摩尔某键时的焓变。 HC1(g-→H(g+C1g);EP=A=431 kJ-mor 2.键能与键解离能(D) (1)键解离能(D)解离气态分子中1摩尔某特定键所需的能量 (②)对双原子分子而言(例HC)其键能数值等于该键的解离能(D), 例:HCI(g H(g) (g) 9=D9=431J ③)双原子分子中若有多个相同的键,则该键的键能为同种键逐级解离能的平 均值。 例:NH分子:键能(E)与键解离能(D)在多原子分子中的区别与关联 NH(g)分子中三个N-H键的键能(E是相同的:三级解离能(D)不同 NH3g)-→NH2(g)+H(g): D=435 kJmol- NH(g)一NH(g)+Hg): D=398Jmol- NHg)-→N(g)+Hg): D=339 kJ-mol- 军a呢a+叹ra+2mH3=435+398+339/3=391 kJ-mol 6-1-2键长 1.键长(L一分子内成键两原子核间的平衡距离。 种键在不同分子中的键长数值基 本上是个定值 例如氢氧键(HO)的键长L0H在不同分子中的数值几乎相等。 H2O H2O2 CH3OH HCOOH 96 97 9696 2.一些双原子分子的键长(表6-1) 表6-1一些双原子分子的键长 键Wpm 键 Lv/pm H-H74.0 H-F 91.3 C1-C1198.8 H-CI 127.4 Br-Br228.4H-C1140.8
第六章 分子的结构与性质 6-1 键参数 6-1-1 键能 1. 键能(E)─— 衡量原子之间形成的化学键强度(键牢固程度)的键参数。粗略而 言,是指在标准状态下气态分子每断裂 1 摩尔某键时的焓变。 HCl(g) ─→ H(g) + Cl(g) ; E θ= = 431kJ·mol-1 2. 键能与键解离能(D) ⑴ 键解离能(D)-解离气态分子中 1 摩尔某特定键所需的能量 ⑵ 对双原子分子而言(例 HCl)其键能数值等于该键的解离能(D)。 例:HCl(g)─→ H(g) + Cl(g); E θ = D θ = 431kJ·mol-1 ⑶双原子分子中若有多个相同的键,则该键的键能为同种键逐级解离能的平 均值。 例:NH3 分子: 键能(E)与键解离能(D) 在多原子分子中的区别与关联 NH3(g)分子中三个 N-H 键的键能(E)是相同的;三级解离能(Di )不同 NH3(g) ─→ NH2(g) + H(g); = 435 kJ·mol- 1 NH2(g) ─→ NH(g) + H(g); = 398 kJ·mol- 1 NH(g) ─→ N(g) + H(g); = 339 kJ·mol- 1 =( + + )/3 =(435+398+339)/3 = 391 kJ·mol-1 6-1-2 键长 1.键长(Lb)──分子内成键两原子核间的平衡距离。 同一种键在不同分子中的键长数值基本上是个定值。 例如氢氧键(H-O)的键长 LO-H在不同分子中的数值几乎相等。 H2O H2O2 CH3OH HCOOH LO-H/p m 96 97 96 96 2.一些双原子分子的键长(表 6-1) 表 6-1 一些双原子分子的键长 键 Lb/pm 键 Lb/pm H-H 74.0 H-F 91.3 Cl-Cl 198.8 H-Cl 127.4 Br-Br 228.4 H-Cl 140.8

-1266.6H-Br160.8 3.两个确定的原子之间,形成的不同的化学键,其键长值越小,键能就越大, 键就越牢固(表6-2)。 表6-2若干化学键的键长和键能 化学键 C-CC-C C=CN-N N=N NN C-NC-NC=N L/pm 154134120146125109.8147132116 E/kJmo356598813160418946285616866 6-1-3键角 键角 分子中两个相邻化学键之间的夹角。键角和键角是描述分子几何结 构的两个要素。例如图6-1中的分子 6-2价键理论 6-2-1共价键 1.共价键的形成 海特勒(Heitler)和伦敦(London)应用量子力学处理两个H原子形成Hz 分子的过程,得到H2分子的能量与核间距离的关系曲线。H2分子基态时的核间 距离d-74pm,小于两个H原子玻尔半径之和(53pm×2=106pm),这表明,在 出分 子中 使两核间形 个H原 子的 道之间发生 成键电子 九道重叠的结果 电子出现的几率密度较大的区域。这样,不仅削弱了两核间 的正电排斥力,而且还增强了核间电子云对两氢核的吸引力,使体系能量得以降 低,从而形成共价键。如图6-2所示。 2.价键理论要点 价健理论(俗称电子配对法)的基本要点是 (1)两原子接近时,自旋方向相反的未成对的价电子可以配对,形成共价键 (2)成键电子的原子轨道如能重登越多,所形成的共价键就越牢固-最大重 原理。 3.共价键的特征 根据价球理论要占 可以推知共价键具有饱和性和方向性 饱和性按要点(1)可推知 ,原子有几个未成对的价电手 般就只能和几个 自旋方向相反的电子配对成键。例如,N原子因为含有三个未成对的价电子,因 此两个N原子间最多只能形成叁键,即形成N=N分子。说明一个原子形成共价 键的能力是有限的,这决定共价键具有饱和性。 希有气体,由于原子没有未成对电子,原子间不成键,因此以单原子分子的 形式存在。但是】 中有些本来成对的价电子, 在特定条件下也有可能被拆为 与成键的例如,硫原子02s2p3s3p的价层 温电负性大的干原子时,价电子对可以拆开,使未成对电子 中原来只有两个未成 个。从而可与6个F原子的未成对电子配对成键,形成SF6分子(结构参见图6-17, 注意!看图后,关闭图6-17的隐藏层,占击刘监器“后退”按朝方可可回到出页)。 方向性按要点(2)可推知 ,形成共价健时,成键电子的原子轨道只有沿着 道伸展的方向进行重叠(s轨道与s轨道重叠例外),才能实现最大限度的重叠
I-I 266.6 H-Br 160.8 3. 两个确定的原子之间,形成的不同的化学键,其键长值越小,键能就越大, 键就越牢固(表 6-2)。 表 6-2 若干化学键的键长和键能 化学键 C-C C=C C≡C N-N N=N N≡N C-N C=N C≡N Lb/pm 154 134 120 146 125 109.8 147 132 116 E θ /(kJ·mol-1 ) 356 598 813 160 418 946 285 616 866 6-1-3 键角 键角-─分子中两个相邻化学键之间的夹角。键角和键角是描述分子几何结 构的两个要素。例如图 6-1 中的分子 6-2 价键理论 6-2-1 共价键 1. 共价键的形成 海特勒(Heitler)和伦敦(London)应用量子力学处理两个 H 原子形成 H2 分子的过程,得到 H2分子的能量与核间距离的关系曲线。H2分子基态时的核间 距离 d=74pm,小于两个 H 原子玻尔半径之和(53pm×2 =106pm),这表明,在 H2分子中,两个 H 原子的 1s 轨道之间发生了重叠。成键电子轨道重叠的结果, 使两核间形成了一个电子出现的几率密度较大的区域。这样,不仅削弱了两核间 的正电排斥力,而且还增强了核间电子云对两氢核的吸引力,使体系能量得以降 低,从而形成共价键。如图 6-2 所示。 2. 价键理论要点 价键理论(俗称电子配对法)的基本要点是: (1) 两原子接近时,自旋方向相反的未成对的价电子可以配对,形成共价键。 (2) 成键电子的原子轨道如能重叠越多,所形成的共价键就越牢固-最大重叠 原理。 3.共价键的特征 根据价键理论要点,可以推知共价键具有饱和性和方向性。 饱和性 按要点(1)可推知,原子有几个未成对的价电子,一般就只能和几个 自旋方向相反的电子配对成键。例如,N 原子因为含有三个未成对的价电子,因 此两个 N 原子间最多只能形成叁键,即形成 N≡N 分子。说明一个原子形成共价 键的能力是有限的,这决定共价键具有饱和性。 希有气体,由于原子没有未成对电子,原子间不成键,因此以单原子分子的 形式存在。但是,原子中有些本来成对的价电子,在特定条件下也有可能被拆为 单电子而参与成键的。例如,硫原子(1s2 2s2 2p6 3s2 3p4 )的价层中原来只有两个未成 对电子。当遇电负性大的 F 原子时,价电子对可以拆开,使未成对电子数增至 6 个。从而可与6个 F 原子的未成对电子配对成键,形成SF6分子(结构参见图 6-17, 注意!看图后,关闭图 6-17 的隐藏层,点击浏览器“后退”按钮方可回到此页)。 方向性 按要点(2)可推知,形成共价键时,成键电子的原子轨道只有沿着轨 道伸展的方向进行重叠(s 轨道与 s 轨道重叠例外),才能实现最大限度的重叠

这就决定了共价键具有方向性, 4.原子轨道的重叠 只有当原子轨道对称性相同的部分重叠,两原子间电子出现的几率密度才会 增大,才能形成化学键称为对称性原则)。以A、B原子的两个原子轨道沿着X 轴方向重叠为例,具体说明之。 (1)当两个原子轨道以对称性相同的部分(即+与“+”,“”与“)相重叠时, 由于原子间电子出现的几率密度比重叠前增大的结果,使两个原子间的结合力大 于两核 的 导致体系能量降低 从而 可能形成共价 这种重 对成键是有效的,称为有效重叠或正重叠。由于原子轨道角度分布突出处往往是 有利于实现最大重叠的地方,所以讨论问题时,常常借用原子轨道角度分布图来 表示原子轨道。图6-3给出原子轨道几种正重叠的示意图。 2)当两个原子轨首以对称性不同部分(即+"与")相重叠时,两原子间由子 出现的几率密度比重叠前减小的结果, 在两原子核之间形成了 系能量升高,难以成键。显然,这种重叠对成键是无效的,称为非有效重叠或负 重叠。图6-4给出原子轨道几种负重叠的示意图。 2 子键 ()离子键的基本概念:1916年德国化学家柯塞尔(W Kossel)提出离子键 的概念。他认为电离能较小的金属原子和电离能较大的非金属原子靠近时,前者 易失去外层电子成正离子,后者易获得电子成正负离子。正负离子之间靠静电引 力结合在 起.生成离子化合物 (②)离子键的本质是正、负离子之间的静电引力。离子键是一种较强的相互 作用力 )离子键可存在于气体分子(例如NaCr离子型分子)内,但大量存在于 离子晶体中。 (4)离子键的特征是既无方向性又无的和性(例如当两个异电荷的离子如 Na和Cr被此吸引形成NaCr离子型分子后,由于离子的电场力无方向性,各自 仍具有吸引异电荷离子的 只要空间条件许可(俗话说"挤得下"),每种离 子均可结合更多的异电荷离子,因此离子键无饱和性。) 2.键型过渡 键型过渡的基本含义两原子的结合是形成共价键还是离子键,取决于两原 子吸引电子能力差别的大小。例如活泼的金属原子和活泼的非金属原子化合成 键,成键电 子有可能完全转移到吸电子能力强的原 从而形成离子 若没有差别,则形成非极性键。因此,从键的极性角度来说,离子键可以看 成是强极性键的极限。 如果把离子键看成是强极性键的极限,把非极性共价键看成是弱极性键的极 限,那么如图6-9所示,极性键可以说是介于非极性键与离子键之间的一种过渡 键型 从表63可看出,成键两元素的电负性差值(△X)越大,键的极性越强 怎样理解化合物中的健性成分?为什么化合物中的化学键绝大部分都是离 子性与共价性兼而有之? 若把非极性键看作纯粹的100%的共价键,把理想中的纯粹的离子键看作为
这就决定了共价键具有方向性。 4. 原子轨道的重叠 只有当原子轨道对称性相同的部分重叠,两原子间电子出现的几率密度才会 增大,才能形成化学键(称为对称性原则)。以 A、B 原子的两个原子轨道沿着 x 轴方向重叠为例,具体说明之。 (1) 当两个原子轨道以对称性相同的部分(即“+”与“+”,“-”与“-”)相重叠时, 由于原子间电子出现的几率密度比重叠前增大的结果,使两个原子间的结合力大 于两核间的排斥力,导致体系能量降低,从而可能形成共价键。显然,这种重叠 对成键是有效的,称为有效重叠或正重叠。由于原子轨道角度分布突出处往往是 有利于实现最大重叠的地方,所以讨论问题时,常常借用原子轨道角度分布图来 表示原子轨道。图 6-3 给出原子轨道几种正重叠的示意图。 (2) 当两个原子轨道以对称性不同部分(即"+"与"-")相重叠时,两原子间电子 出现的几率密度比重叠前减小的结果,在两原子核之间形成了一个垂直于 x 轴 的、电子的几率密度几乎等于零的平面(称节面),由于核间排斥力占优势,使体 系能量升高,难以成键。显然,这种重叠对成键是无效的,称为非有效重叠或负 重叠。图 6-4 给出原子轨道几种负重叠的示意图。 6-2-2 离子键 1. 离子键 ⑴ 离子键的基本概念:1916 年德国化学家柯塞尔(W Kossel)提出离子键 的概念。他认为电离能较小的金属原子和电离能较大的非金属原子靠近时,前者 易失去外层电子成正离子,后者易获得电子成正负离子。正负离子之间靠静电引 力结合在一起,生成离子化合物。 ⑵ 离子键的本质是正﹑负离子之间的静电引力。离子键是一种较强的相互 作用力。 ⑶ 离子键可存在于气体分子(例如 Na+Cl-离子型分子)内,但大量存在于 离子晶体中。 ⑷ 离子键的特征是既无方向性又无饱和性(例如当两个异电荷的离子如 Na+和 Cl-彼此吸引形成 Na+Cl-离子型分子后,由于离子的电场力无方向性,各自 仍具有吸引异电荷离子的能力,只要空间条件许可(俗话说"挤得下"),每种离 子均可结合更多的异电荷离子,因此离子键无饱和性。) 2.键型过渡 键型过渡的基本含义 两原子的结合是形成共价键还是离子键,取决于两原 子吸引电子能力差别的大小。例如 活泼的金属原子和活泼的非金属原子化合成 键,成键电子有可能完全转移到吸电子能力强的原子上去,从而形成离子键;反 之,若没有差别,则形成非极性键。因此,从键的极性角度来说,离子键可以看 成是强极性键的极限。 如果把离子键看成是强极性键的极限,把非极性共价键看成是弱极性键的极 限,那么如图 6-9 所示,极性键可以说是介于非极性键与离子键之间的一种过渡 键型。 从表 6-3 可看出,成键两元素的电负性差值(Δχ)越大,键的极性越强。 怎样理解化合物中的键性成分? 为什么化合物中的化学键绝大部分都是离 子性与共价性兼而有之? 若把非极性键看作纯粹的 100%的共价键,把理想中的纯粹的离子键看作为

100%的离子键,那么,从键型过渡的角度来说,极性共价键又可以看作为含有 小部分离子键成分和大部分共价键成分的中间类型的化学键。当极性键向离子键 过渡时,共价健成分(又称共价性)逐渐 少 而离子键成分( (称离子性 渐增加。因此,从这个意义上来说,绝大多数的离子键都不是典型的,只是离子 性占优势而已。 6-3分子的几何构型 63-1价键理论的局限性 价键理论的局限性 按照价键理论,C原子最多只能形成四个键,且用s轨道与用p轨道形成的 键应有不同,经实验测知,CH分子的空间结构如图6-10所示。 显然,轨道重叠,电子配对的价键理论不足以解释一般多原子分子的价键形 成(基态C原子的价层电子构型是2s22印:为什么只有两个未成对电子,实际中 却可以形成四个 一H键 CH分子中基态C原子的价层电子构型是2s22p2。 按照这个结构,C原子只能提供两个未成对电子,与H原子形成两个C H键! 若虑到C原子价层有一个空的2n轨首,日能量比2s轨首只销高一些,如 果设想在成键时有 个2s电子会被激发到2p的 一个空轨道上去,而使价层内具 有四个未成对电子,这样,可以和H原子形成四个C一H健。因为从能量的观 点来说,2s电子被微发到2p所需要的能量,可以被形成四个C一H键后放出的 能量补偿而有余)和几何构型(按价键理论推测,甲烷的化学键特点 用烷的四个C一H键将是不完全等同的: 由于2D轨道较2s轨道角度分布有一突出的部分,和相邻原子轨道重叠较 大 些,因而由p电子构成的C-H键其键能理应较大 一些,而由s电子所构成 的C一H键其键能理应较小一些;由p电子所构成的三个C一H键理应互相垂直。) 问题。 63-2杂化轨道理论 1.杂化轨道理论的简要内容 1)在形成分子(主要是化合物)时,同一中若干能景相折的陌子轨首 (一般为同一能级组的原子轨道)相互叠加(杂化)形成一组同样数目的新的原 子轨道。 有关概念: 杂化一轨道的相互叠加过程叫原子轨道的杂化。 杂化轨道一原子轨道叠加后产生的新的原子轨道叫杂化轨道。 (②)杂化轨道比原来的轨道成键能力强, 形成的化学键键能大 使生成的分 子更稳定。由于成键原子轨道杂化后,轨道角度分布图的形状发生了变化(形光 是一头大,一头小),杂化轨道在某些方向上的角度分布,比未杂化的P轨道和 s轨道的角度分布大得多,它的大头在成键时与原来的轨道相比能够形成更大的 重叠,因此杂化轨道比原有的原子轨道成键能力更强。 (3)形成的杂化轨道之间应尽可能地满足最小排斥原理(化学键间排斥力越
100%的离子键,那么,从键型过渡的角度来说,极性共价键又可以看作为含有 小部分离子键成分和大部分共价键成分的中间类型的化学键。当极性键向离子键 过渡时,共价键成分(又称共价性)逐渐减少,而离子键成分(又称离子性)逐 渐增加。因此,从这个意义上来说,绝大多数的离子键都不是典型的,只是离子 性占优势而已。 6-3 分子的几何构型 6-3-1 价键理论的局限性 价键理论的局限性 按照价键理论,C 原子最多只能形成四个键,且用 s 轨道与用 p 轨道形成的 键应有不同,经实验测知,CH4分子的空间结构如图 6-10 所示。 显然,轨道重叠,电子配对的价键理论不足以解释一般多原子分子的价键形 成(基态 C 原子的价层电子构型是 2s2 2p2 :为什么只有两个未成对电子,实际中 却可以形成四个 C-H 键? CH4分子中基态 C 原子的价层电子构型是 2s2 2p2。 按照这个结构,C 原子只能提供两个未成对电子,与 H 原子形成两个 C- H 键; 考虑到 C 原子价层有一个空的 2p 轨道,且能量比 2s 轨道只稍高一些,如 果设想在成键时有一个 2s 电子会被激发到 2p 的一个空轨道上去,而使价层内具 有四个未成对电子,这样,可以和 H 原子形成四个 C-H 键。因为从能量的观 点来说,2s 电子被激发到 2p 所需要的能量,可以被形成四个 C-H 键后放出的 能量补偿而有余)和几何构型(按价键理论推测,甲烷的化学键特点 甲烷的四个 C-H 键将是不完全等同的: 由于 2p 轨道较 2s 轨道角度分布有一突出的部分,和相邻原子轨道重叠较 大一些,因而由 p 电子构成的 C-H 键其键能理应较大一些,而由 s 电子所构成 的C-H 键其键能理应较小一些;由p电子所构成的三个C-H键理应互相垂直。) 问题。 6-3-2 杂化轨道理论 1. 杂化轨道理论的简要内容 ⑴ 在形成分子(主要是化合物)时,同一原子中若干能量相近的原子轨道 (一般为同一能级组的原子轨道) 相互叠加(杂化)形成一组 同样数目的新的原 子轨道。 有关概念: 杂化 ── 轨道的相互叠加过程叫原子轨道的杂化。 杂化轨道──原子轨道叠加后产生的新的原子轨道叫杂化轨道。 ⑵ 杂化轨道比原来的轨道成键能力强,形成的化学键键能大,使生成的分 子更稳定。由于成键原子轨道杂化后,轨道角度分布图的形状发生了变化(形状 是一头大,一头小),杂化轨道在某些方向上的角度分布,比未杂化的 p 轨道和 s 轨道的角度分布大得多,它的大头在成键时与原来的轨道相比能够形成更大的 重叠,因此杂化轨道比原有的原子轨道成键能力更强。 ⑶ 形成的杂化轨道之间应尽可能地满足最小排斥原理(化学键间排斥力越
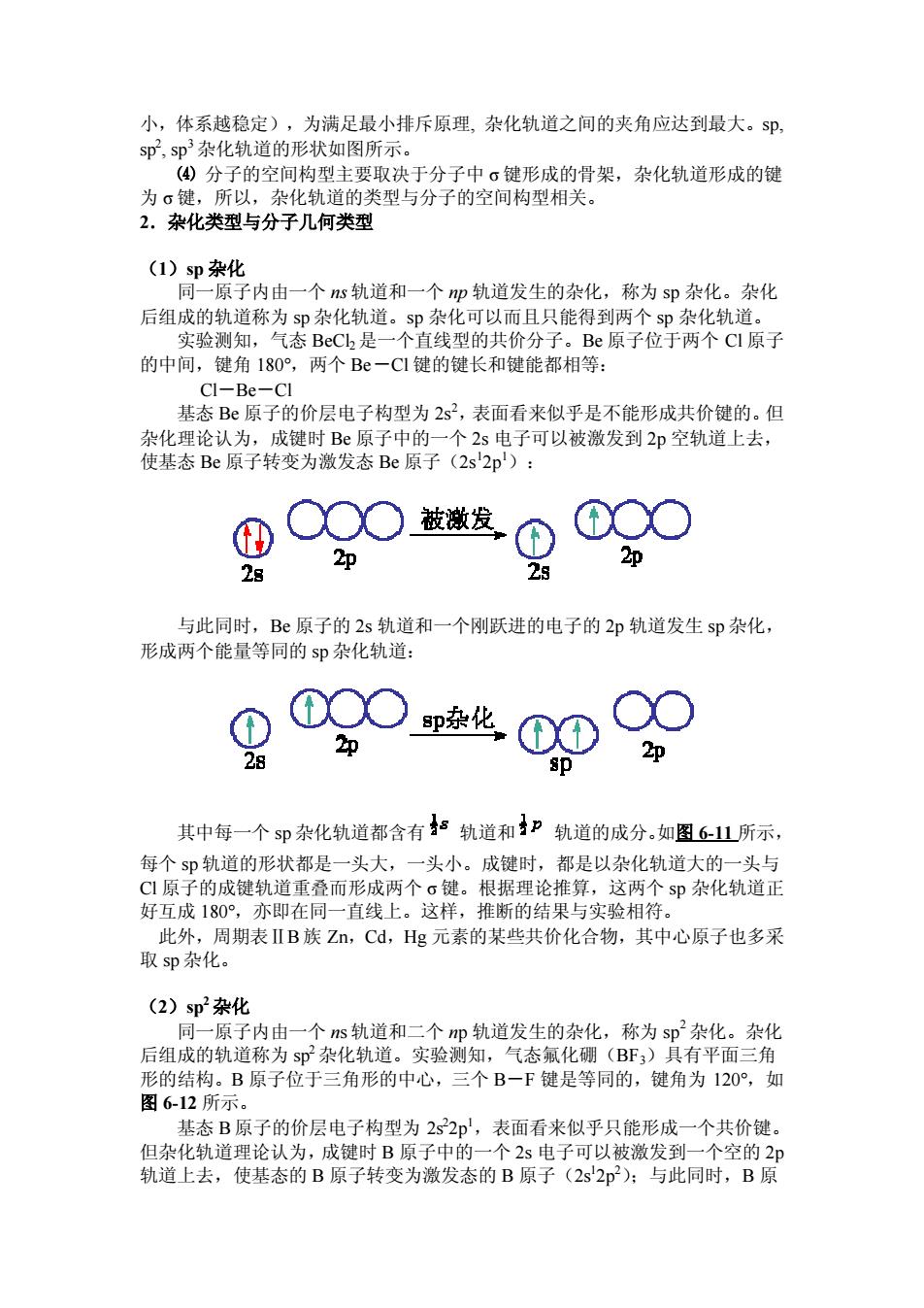
小,体系越稳定),为满足最小排斥原理,杂化轨道之间的夹角应达到最大。5即, sp,sp杂化轨道的形状如图所示 (④分子的空间构型主要取决于分子中。健形成的骨架,杂化轨道形成的键 为。键,所以,杂化轨道的类型与分子的空间构型相关。 2.杂化类型与分子几何类型 (1)sD杂化 同一原子内由一个s轨道和一个即轨道发生的杂化,称为sD杂化。杂化 可以而且只能得到两 sp杂化轨道。 的中间,键角180°,两个Be一C1键的键长和键能都相等: CI-Be-CI 基态B原子的价层电子构型为2s?,表面看来似乎是不能形成共价键的。但 杂化理论认为,成键时B原子中的一个2s电子可以被激发到2p空轨道上去 使基态Be原子转变为激发态Be原子(2s'2p'): 0○OO被漾发 ①O0 2p 2p 28 2 与此同时,B原子的2s轨道和一个刚跃进的电子的2p轨道发生sp杂化, 形成两个能量等同的sp杂化轨道: ①O○p化0① 2p 2p 其中每一个sP杂化轨道都含有”轨道和”轨道的成分。如图61山所示, 每个$即轨道的形状都是一头大,一头小。成键时,都是以杂化轨道大的一头与 C1原子的成键轨道重叠而形成两个σ键。根据理论推算,这两个sp杂化轨道正 好互成180°,亦即在同一直线上。这样,推断的结果与实验相符。 此外,周期表ⅡB族Zn,Cd,Hg元素的某些共价化合物,其中心原子也多采 取sp杂化。 (2)sp2杂化 同一原子内由一个s轨道和二个m轨道发生的杂化,称为sp杂化。杂化 后组成的轨道称为sp杂化轨道。实验测知,气态氟化硼(BF;)具有平面三角 形的结构。B原子位于三角形的中心,三个B一F键是等同的,键角为120,如 图6-12所示 基态B原子的价层电子构型为2s22印,表面看来似乎只能形成一个共价键。 但杂化轨道理论认为,成键时B原子中的一个2s电子可以被激发到一个空的2p 轨道上去,使基态的B原子转变为激发态的B原子(2s2p):与此同时,B原
小,体系越稳定),为满足最小排斥原理, 杂化轨道之间的夹角应达到最大。sp, sp2 , sp3杂化轨道的形状如图所示。 ⑷ 分子的空间构型主要取决于分子中 σ键形成的骨架,杂化轨道形成的键 为 σ键,所以,杂化轨道的类型与分子的空间构型相关。 2.杂化类型与分子几何类型 (1)sp 杂化 同一原子内由一个 ns 轨道和一个 np 轨道发生的杂化,称为 sp 杂化。杂化 后组成的轨道称为 sp 杂化轨道。sp 杂化可以而且只能得到两个 sp 杂化轨道。 实验测知,气态 BeCl2是一个直线型的共价分子。Be 原子位于两个 Cl 原子 的中间,键角 180°,两个 Be-Cl 键的键长和键能都相等: Cl-Be-Cl 基态 Be 原子的价层电子构型为 2s2,表面看来似乎是不能形成共价键的。但 杂化理论认为,成键时 Be 原子中的一个 2s 电子可以被激发到 2p 空轨道上去, 使基态 Be 原子转变为激发态 Be 原子(2s1 2p1): 与此同时,Be 原子的 2s 轨道和一个刚跃进的电子的 2p 轨道发生 sp 杂化, 形成两个能量等同的 sp 杂化轨道: 其中每一个 sp 杂化轨道都含有 轨道和 轨道的成分。如图 6-11 所示, 每个 sp 轨道的形状都是一头大,一头小。成键时,都是以杂化轨道大的一头与 Cl 原子的成键轨道重叠而形成两个 σ键。根据理论推算,这两个 sp 杂化轨道正 好互成 180°,亦即在同一直线上。这样,推断的结果与实验相符。 此外,周期表ⅡB族 Zn,Cd,Hg 元素的某些共价化合物,其中心原子也多采 取 sp 杂化。 (2)sp2杂化 同一原子内由一个 ns 轨道和二个 np 轨道发生的杂化,称为 sp 2杂化。杂化 后组成的轨道称为 sp2杂化轨道。实验测知,气态氟化硼(BF3)具有平面三角 形的结构。B 原子位于三角形的中心,三个 B-F 键是等同的,键角为 120°,如 图 6-12 所示。 基态 B原子的价层电子构型为 2s2 2p1,表面看来似乎只能形成一个共价键。 但杂化轨道理论认为,成键时 B 原子中的一个 2s 电子可以被激发到一个空的 2p 轨道上去,使基态的 B 原子转变为激发态的 B 原子(2s1 2p2);与此同时,B 原