
否则空气中的氧将I一氧化的速度也要加快,给测定带来误差。 对大多数反应来说,升高溶液的温度可加快反应速率。通常溶液温度每升高O℃,万应 率约增大2~3倍。例如,在酸性溶液中MO,与C0:的反阿反应式(6一13)],在室 温下,反应速率缓慢。如果将溶液加热,反应速率便大为加快。所以用KMO,滴定H:C2O 时,通常将溶液加热至5~85。但升高温度时还应考虑到其他一些可能引起的不利因素。 对于反应式(6-一13温度过高,会引起部分HC,0分解.有些物质〔例如)较易挥发 如将溶液加热,则会 引起挥发损失,所以对于反应 不能用加热的 办法来 提高 反应速率。又如有些物质(例如Sn、Fe等)很容易被空气中的氧所氧化,如将溶液加 热,就会促使它们氧化,从而引起误差。只有采用别的办法提高反应速率。 3.催化剂 氧化还原反应中经常利用催化剂来改变反应速率。催化剂可分为正催化剂和负催化剂。正催 化剂加快反应速率 负催化剂减慢反应速率 催化反应的机理非常复杂。例如上述MO:与CO之间的反应 ,Mn2的存在能催化反应 迅速进行。其反应机理可能是,在C2O,存在下MnO:被Mn氧化而生成Mn(Ⅲ): Mn0,-+4Mn2+5nC,0,-+8H→5Mn(C,0).B2+4H,0 上述反应是分步进行的,反应过程可简单表示如下: Mn(M)Mn( n(Mn() 一MnN)+Mn(Ⅲ) →Mn(Ⅲ) Mn()Mn(C.Mn(I)+CO 在此,M参加反应的中间步骤,加速了反应,但在最后又重新产生出来, 它起了催化剂 的作用。同时,在反应式(6一13)中M是反应的生成物之一,因此假如在溶液中并不另 外加人二价的锰盐,则在反应开始时由于一般KMO:溶液中Mn“含量极少,所以虽加热到 7585℃,反应进行得仍较为缓慢,MO,褪色很慢。但反应一经开始,溶液中产生了少量 的Mm后,由于Mn+的催化作用,就使以后的反应大力加速。这里加速反应的催化剂M 是由反应本身生成的,因此这种反应称为自动催化作用。 能斯特方程式联立求得。 令化学计量点时的电极电位为中s即,则
否则空气中的氧将 I - 氧化的速度也要加快,给测定带来误差。 2.温度 对大多数反应来说,升高溶液的温度可加快反应速率。通常溶液温度每升高 10℃,反应速 率约增大 2~3倍。例如,在酸性溶液中 MnO4 -与 C2O4 2-的反应[反应式(6—13)],在室 温下,反应速率缓慢。如果将溶液加热,反应速率便大为加快。所以用 KMnO4 滴定 H2C2O4 时,通常将溶液加热至 75~85 t。但升高温度时还应考虑到其他一些可能引起的不利因素。 对于反应式(6—13),温度过高,会引起部分 H2C2O4 分解。有些物质(例如 I2)较易挥发, 如将溶液加热,则会引起挥发损失,所以对于反应式(6—14),不能用加热的办法来提高其 反应速率。又如有些物质(例如 Sn2+、Fe2+等)很容易被空气中的氧所氧化,如将溶液加 热,就会促使它们氧化,从而引起误差。只有采用别的办法提高反应速率。 3.催化剂 氧化还原反应中经常利用催化剂来改变反应速率。催化剂可分为正催化剂和负催化剂。正催 化剂加快反应速率,负催化剂减慢反应速率。 催化反应的机理非常复杂。例如上述 MnO4 -与 C2O4 2- 之间的反应,Mn2+的存在能催化反应 迅速进行。其反应机理可能是,在 C2O4 2-存在下 MnO4 -被 Mn 2+氧化而生成 Mn(Ⅲ): MnO4 -+4 Mn2++5nC2O4 2-+8H+→5Mn(C2O4)n (3-2n)+4H2O 上述反应是分步进行的,反应过程可简单表示如下: 在此,Mn2+参加反应的中间步骤,加速了反应,但在最后又重新产生出来,它起了催化剂 的作用。同时,在反应式(6—13)中 Mn2+是反应的生成物之一,因此假如在溶液中并不另 外加人二价的锰盐,则在反应开始时由于一般 KMnO4溶液中 Mn2+含量极少,所以虽加热到 75~85℃,反应进行得仍较为缓慢,MnO4 -褪色很慢。但反应一经开始,溶液中产生了少量 的 Mn2+后,由于 Mn2+的催化作用,就使以后的反应大力加速。这里加速反应的催化剂 Mn2+ 是由反应本身生成的,因此这种反应称为自动催化作用。 能斯特方程式联立求得。 令化学计量点时的电极电位为φsp,则

pp=p是c+0.059gcN ce(I】 =2+059 (6-15) 又令 =0 则由式(6-15)可得 +0.059 lg CD2 Ce(I 9w+009k 将上两式相加得 2,-p+9+0.09 根据前述滴定反应式,当加人Ce(SO)2的物质的量与会F®2+的物质的量相等时 Ccev=CeⅡCCey=CFam.此时 -0 故 e=+2 9w=0.6V1.4y=1.06v 对于一般的可逆对称氧化还原反应 n0+nRad一n,Red+n10 可用类似方法求得,化学计量点时的电位中s即与中1°,中2°的关系 (6-16) 月1十2 如果电对的氧化态和还原态的系数不等,即不对称,例如 Cr,0号+6Fe2+14H*2C23+6Fe+7H20 则中即除与电⊙及n有关外,还和离子的浓度有关。 3.化学计量点后 此时可利用Ce“/Ce电对米计算电位值。例如,当加人过量0.1%Ce“时, Cce (IV yCce(lI=1/103,故
根据前述滴定反应式,当加人 Ce(SO4)2 的物质的量与会 Fe2+的物质的量相等时, CCe(Ⅳ)=CFe(Ⅱ),CCe(Ⅲ)=CFe(Ⅲ),此时 对于一般的可逆对称氧化还原反应 可用类似方法求得,化学计量点时的电位φsp 与φ1 ○- ′ ,φ2 ○- ′ 的关系 如果电对的氧化态和还原态的系数不等,即不对称,例如 则φsp 除与φ ○- ′ 及 n 有关外,还和离子的浓度有关。 3.化学计量点后 此时可利用 Ce4+/Ce3+电对来计算电位值 。例如,当加人过量 0.1% Ce4+时, CCe(Ⅳ)/CCe(Ⅲ)=1/103 ,故
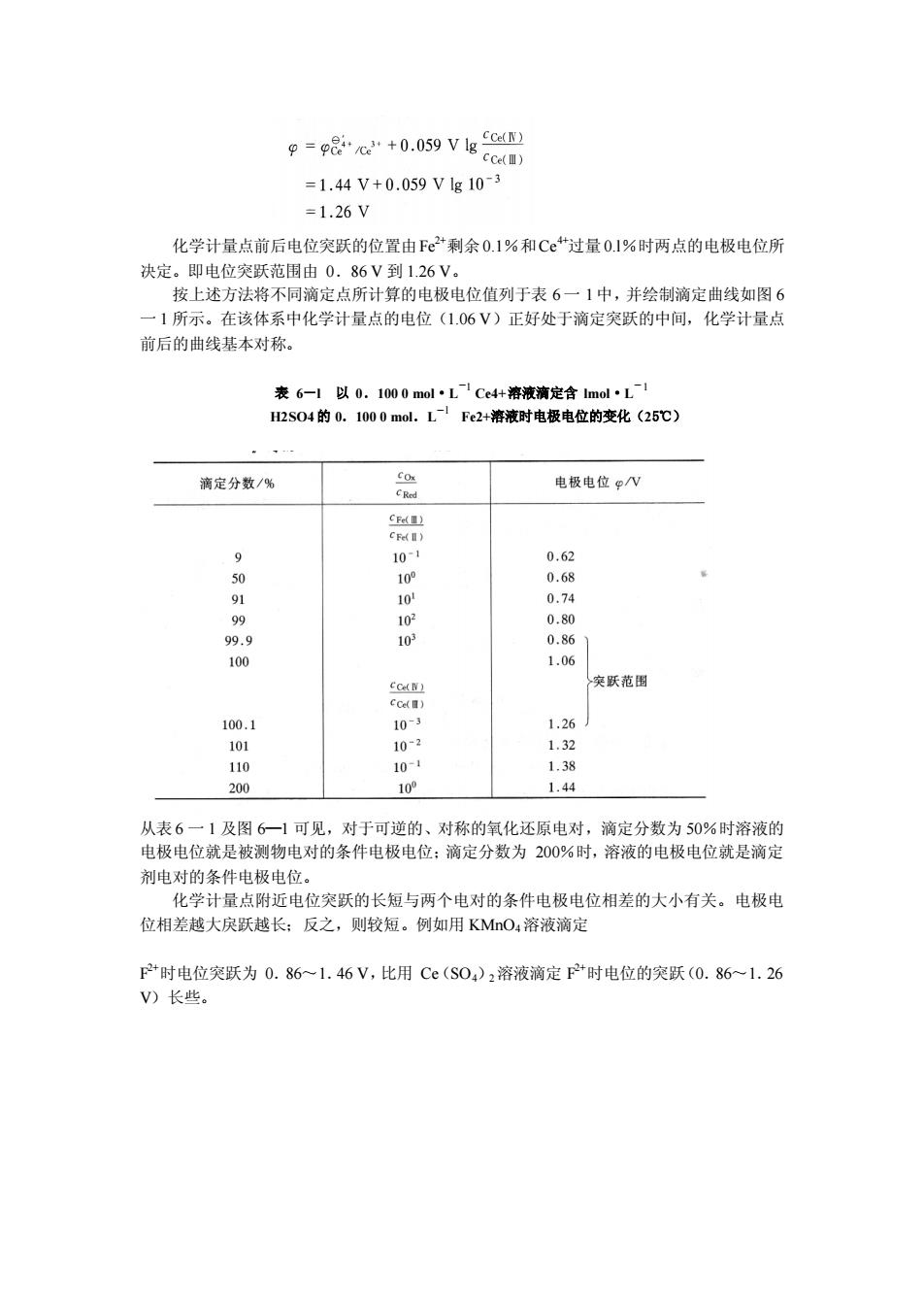
p=e+059ve2 =1.44V+0.059Vg103 =1.26V 化学计量点前后电位突跃的位置由Fe剩余0.1%和C“过量0.1%时两点的电极电位所 1所示。在该体系中化学计量点的电位(1.06V)正好处于滴定突跃的中间,化学计量点 前后的曲线基本对称。 表6一以0.1000mol·LCe4+液清定含mlL1 H2S04的0. +落液时电极 电位的变化(25℃) 滴定分数/% 电极电位N 10-1 0.62 0.68 9 10 0.74 突跃范围 8 1.26 1.32 o 0- 1.38 200 10P 1.44 从表6 1及图6一1可见 对于可逆的、对称的氧化还原电对,滴定分数为50%时溶液的 电极电位就是被物电对的条件电极电位:滴定分数为200%时,溶液的电极电位就是滴定 剂电对的条件电极电位。 化学计量点附近电位突跃的长短与两个电对的条件电极电位相差的大小有关。电极电 位相差越大戾跃越长: 反之,则较短。例如用KMnOa溶液滴定 F时电位突跃为0.86一1.46V,比用C(S0)2溶液滴定F2+时电位的突跃(0.86一1.26 V)长些
化学计量点前后电位突跃的位置由Fe2+剩余 0.1%和Ce4+过量 0.l%时两点的电极电位所 决定。即电位突跃范围由 0.86 V 到 1.26 V。 按上述方法将不同滴定点所计算的电极电位值列于表 6一 1中,并绘制滴定曲线如图 6 一 1 所示。在该体系中化学计量点的电位(1.06 V)正好处于滴定突跃的中间,化学计量点 前后的曲线基本对称。 表 6-l 以 0.100 0 mol·L -1 Ce4+溶液滴定含 lmol·L -1 H2SO4 的 0.100 0 mol.L -l Fe2+溶液时电极电位的变化(25℃) 从表 6 一 1 及图 6—1 可见,对于可逆的、对称的氧化还原电对,滴定分数为 50%时溶液的 电极电位就是被测物电对的条件电极电位;滴定分数为 200%时,溶液的电极电位就是滴定 剂电对的条件电极电位。 化学计量点附近电位突跃的长短与两个电对的条件电极电位相差的大小有关。电极电 位相差越大戾跃越长;反之,则较短。例如用 KMnO4溶液滴定 F 2+时电位突跃为 0.86~1.46 V,比用 Ce(SO4)2溶液滴定 F 2+时电位的突跃(0.86~1.26 V)长些
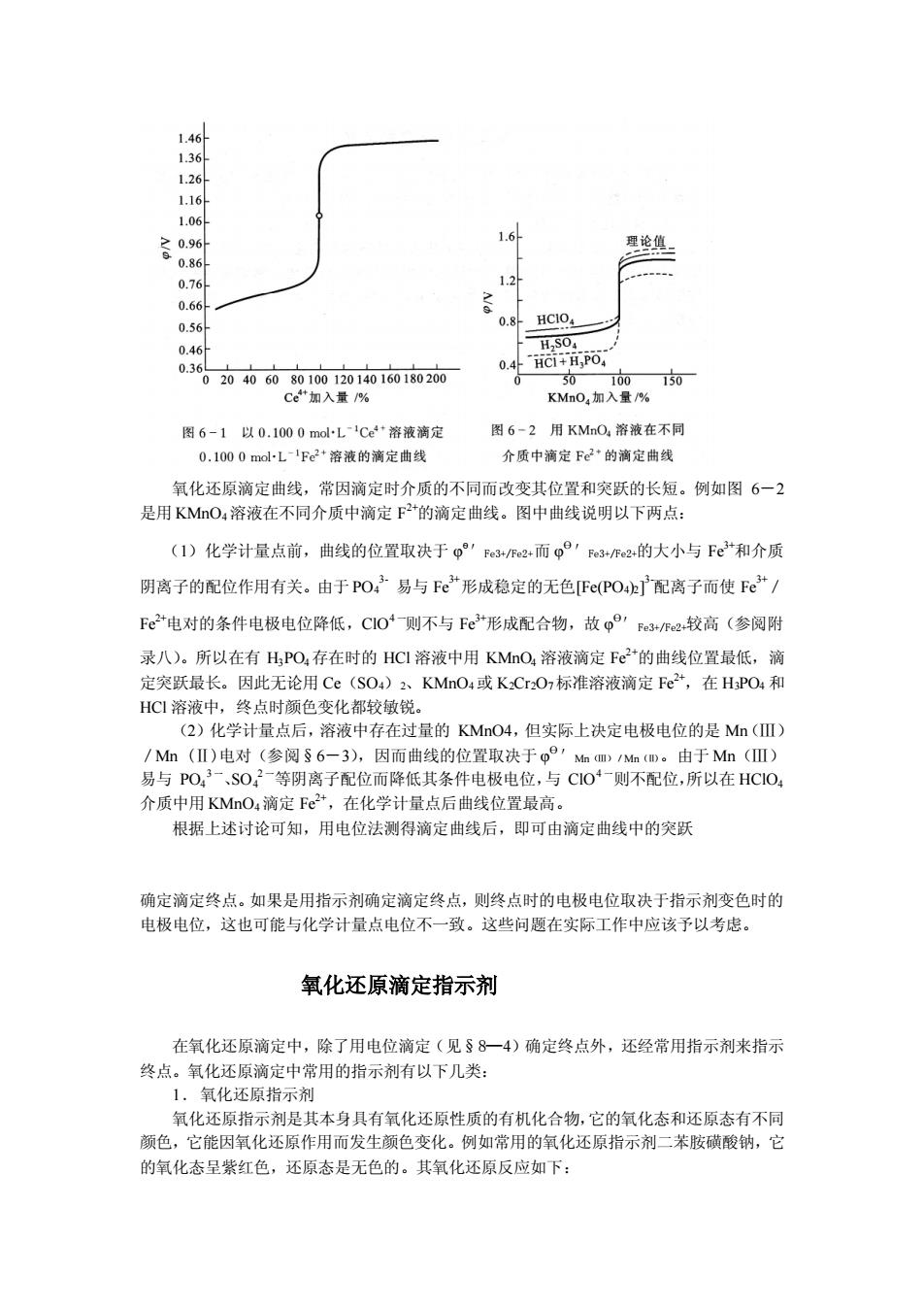
126 026 0.8 HC1O 50 Ce入量% 图6-1以0.1000maL1Ce“游液滴定 图6-2用KMO,溶液在不同 0.1000malL-'pe2*溶液的滴定曲线 介质中滴定F+的滴定曲线 氧化还原滴定曲线,常因滴定时介质的不同而改变其位置和突跃的长短。例如图6一2 是用KMO,溶液在不同介质中滴定F2+的滴定曲线。图中曲线说明以下两点: (1)化学计量点前,曲线的位置取决于Fe3/2而93作2的大小与Fe和介质 阴离子的配位作用有关。由于PO:易与Fe”形成稳定的无色Fe(PO.E]配离子而使Fe/ F©电对的条件电极电位降低,CI0-则不与Fe形成配合物,故p°'e+R2较高(参阅附 录八)。所以在有H,PO,存在时的HCI溶液中用KMO,溶液滴定Fe+的曲线位置最低,滴 定突跃最长。因此无论用Ce(SO)、KMnO4或K,Cr,O,标准溶液滴定Fe2,在HPO和 HC溶液中, 终点 色变化都较敏锐 (2)化学计量点后,溶液中存在过量的KMO4,但实际上决定电极电位的是Mn(Ⅲ) /Mn(Ⅱ)电对(参阅§6一3),因而曲线的位置取决于p,mMD。由于Mn(Ⅲ) 易与PO,3一S0,2一等阴离子配位而降低其条件电极电位,与C104一则不配位,所以在HC1O, 介历中用KMnO,滴定Fe,在化学计量点后曲线位置最高。 根据上述讨论可知,用电位法测得滴定曲线后,即可由滴定曲线中的突跃 确定滴定终点。如果是用指示剂确定滴定终点,则终点时的电极电位取决于指示剂变色时的 电极电位,这也可能与化学计量点电位不一致。这些问题在实际工作中应该予以考虑。 氧化还原滴定指示剂 在氧化还原滴定中,除了用电位滴定(见§8一4)确定终点外,还经常用指示剂来指示 终点。氧化还原滴定中常用的指示剂有以下几类: 化还原封 氧化还原指示剂是其本身具有氧化还原性质的有机化合物,它的氧化态和还原态有不同 颜色,它能因氧化还原作用而发生颜色变化。例如常用的氧化还原指示剂二苯胺磺酸钠,它 的氧化态呈紫红色,还原态是无色的。其氧化还原反应如下:
氧化还原滴定曲线,常因滴定时介质的不同而改变其位置和突跃的长短。例如图 6-2 是用 KMnO4溶液在不同介质中滴定 F 2+的滴定曲线。图中曲线说明以下两点: (1)化学计量点前,曲线的位置取决于 φ Ө′Fe3+/Fe2+而 φ Ө′Fe3+/Fe2+的大小与 Fe3+和介质 阴离子的配位作用有关。由于 PO4 3- 易与 Fe3+形成稳定的无色[Fe(PO4)2] 3-配离子而使 Fe3+/ Fe2+电对的条件电极电位降低,ClO4 一则不与 Fe3+形成配合物,故 φ Ө′Fe3+/Fe2+较高(参阅附 录八)。所以在有 H3PO4存在时的 HCl 溶液中用 KMnO4 溶液滴定 Fe2+的曲线位置最低,滴 定突跃最长。因此无论用 Ce(SO4)2、KMnO4或 K2Cr2O7标准溶液滴定 Fe2+,在 H3PO4 和 HCl 溶液中,终点时颜色变化都较敏锐。 (2)化学计量点后,溶液中存在过量的 KMnO4,但实际上决定电极电位的是 Mn(Ⅲ) /Mn (Ⅱ)电对(参阅§6-3),因而曲线的位置取决于 φ Ө′Mn(Ⅲ)/Mn (Ⅱ)。由于 Mn(Ⅲ) 易与 PO4 3 一、SO4 2 一等阴离子配位而降低其条件电极电位,与 ClO4 一则不配位,所以在 HClO4 介质中用 KMnO4滴定 Fe2+,在化学计量点后曲线位置最高。 根据上述讨论可知,用电位法测得滴定曲线后,即可由滴定曲线中的突跃 确定滴定终点。如果是用指示剂确定滴定终点,则终点时的电极电位取决于指示剂变色时的 电极电位,这也可能与化学计量点电位不一致。这些问题在实际工作中应该予以考虑。 氧化还原滴定指示剂 在氧化还原滴定中,除了用电位滴定(见§8—4)确定终点外,还经常用指示剂来指示 终点。氧化还原滴定中常用的指示剂有以下几类: 1.氧化还原指示剂 氧化还原指示剂是其本身具有氧化还原性质的有机化合物,它的氧化态和还原态有不同 颜色,它能因氧化还原作用而发生颜色变化。例如常用的氧化还原指示剂二苯胺磺酸钠,它 的氧化态呈紫红色,还原态是无色的。其氧化还原反应如下: