
植物食品的蛋白质含量测定,但因样品中常含有核酸、生物碱、含氮类脂、卟啉以及含氮氨色 素等非蛋白质的含氨化合物,故结果称为相蛋白质含量 量法、自动定氯仪 对前三种方法予以介绍。 1.常量凯氏定氨法 (1)原理 样品与浓硫酸和催化剂一同加热消化,使蛋白质分解,其中碳和氢被氧化成二氧化碳 和水逸出,而样品中的有机氮转化为氨与硫酸结合成硫酸铵。然后加碱蒸馏,使氨蒸出,用 硼酸吸收后再以标准盐酸或硫酸溶液滴定。根据标准酸消耗量可计算出蛋白质的含量。 ①样品消化:消化反应方程式如下 2NH (CH)COOH+13HSO =(NH),S01+6C02+12S0.+16H0 浓硫酸具有脱水性,使有机物脱水后被炭化为碳、氢、氮。 浓酸又有氧化性,将有机物炭化后的碳成为二氧化碳,硫酸则被还原成二氧化硫: 2H2S04+C= =2S02+2H,0+C021 二氧化硫使氨还原为氨,本身侧被氧化为三氧化硫,氨随之与硫酸作用生成硫酸铵留在 酸性溶液中: =(NH4)2S0 在消化反应中,为了加速蛋白质的分解,缩短消化时间,常加入下列物质: 1)硫酸钾加入硫酸钾可以提高溶液的沸点而加快有机物分解。它与硫酸作用生成 硫酸氢钾可提高反应温度,一般纯硫酸的沸点在340℃左右,而添加硫酸钾后,可使温度提 高至400℃以上,原因主要在于随者消化过程中硫酸不新地被分解,水分不断逸出而使硫酸 钾浓度增大,故沸点升高, 其反应式如下 K2S04+H2S04== =2KHSO4 2KHSO= =KS04+H,0↑+S03 但硫酸钾加入最不能太大,否则消化体系温度过高,又会引起己生成的铵盐发生热分解放出 氨而造成损失: (NH)2SO4- NH3↑+(NH)HSO △ (NH)HSO →NH↑+S01+H,0 除硫酸钾外,也可以加入硫酸钠、氯化钾等盐类来提高沸点,但效果不如硫酸钾 2)硫酸铜CuSO4 硫酸铜起催化剂的作用。凯氏定氮法中可用的催化剂种类很多 除硫酸铜外,还有氧化汞、汞、硒粉、二氧化钛等,但考虑到效果、价格及环境污染等多种 因素,应用最广泛的是硫酸铜、使用时常加入少量过氧化氢、次氯酸钾等作为氧化剂以加速 有机物氧化,硫酸铜的作用机理如下所示: 20s0,4 3CuS04+S02↑+02
植物食品的蛋白质含量测定,但因样品中常含有核酸、生物碱、含氮类脂、卟啉以及含氮色 素等非蛋白质的含氮化合物,故结果称为粗蛋白质含量。 凯氏定氮法由 Kieldahl 于 1833 年首先提出,经过长期改进,迄今已演变成常量法、微 量法、自动定氮仪法、半微量法及改良凯氏法等多种,至今仍被作为标准检验方法。下面仅 对前三种方法予以介绍。 1.常量凯氏定氮法 (1)原理 样品与浓硫酸和催化剂一同加热消化,使蛋白质分解,其中碳和氢被氧化成二氧化碳 和水逸出,而样品中的有机氮转化为氨与硫酸结合成硫酸铵。然后加碱蒸馏,使氨蒸出,用 硼酸吸收后再以标准盐酸或硫酸溶液滴定。根据标准酸消耗量可计算出蛋白质的含量。 ①样品消化:消化反应方程式如下 2NH3(CH2)2COOH + 13H2SO4 =====(NH4)2SO4 + 6CO2 + 12SO2 + 16H2O 浓硫酸具有脱水性,使有机物脱水后被炭化为碳、氢、氮。 浓硫酸又有氧化性,将有机物炭化后的碳成为二氧化碳,硫酸则被还原成二氧化硫: 2H2SO4 + C ===== 2SO2 + 2H2O + CO2↑ 二氧化硫使氮还原为氨,本身则被氧化为三氧化硫,氨随之与硫酸作用生成硫酸铵留在 酸性溶液中: H2SO4 + 2NH3 =====(NH4)2SO4 在消化反应中,为了加速蛋白质的分解,缩短消化时间,常加入下列物质: 1)硫酸钾 加入硫酸钾可以提高溶液的沸点而加快有机物分解。它与硫酸作用生成 硫酸氢钾可提高反应温度,一般纯硫酸的沸点在 340℃左右,而添加硫酸钾后,可使温度提 高至 400℃以上,原因主要在于随着消化过程中硫酸不断地被分解,水分不断逸出而使硫酸 钾浓度增大,故沸点升高,其反应式如下: K2SO4 + H2SO4 ===== 2KHSO4 2KHSO4 ===== K2SO4 + H2O↑+ SO3 但硫酸钾加入量不能太大,否则消化体系温度过高,又会引起已生成的铵盐发生热分解放出 氨而造成损失: (NH4)2SO4 NH3↑+(NH4)HSO4 (NH4)HSO4 NH3↑+ SO3↑+ H2O 除硫酸钾外,也可以加入硫酸钠、氯化钾等盐类来提高沸点,但效果不如硫酸钾。 2)硫酸铜 CuSO4 硫酸铜起催化剂的作用。凯氏定氮法中可用的催化剂种类很多, 除硫酸铜外,还有氧化汞、汞、硒粉、二氧化钛等,但考虑到效果、价格及环境污染等多种 因素,应用最广泛的是硫酸铜、使用时常加入少量过氧化氢、次氯酸钾等作为氧化剂以加速 有机物氧化,硫酸铜的作用机理如下所示: 2CuSO4 Cu2SO4 + SO2↑+ O2
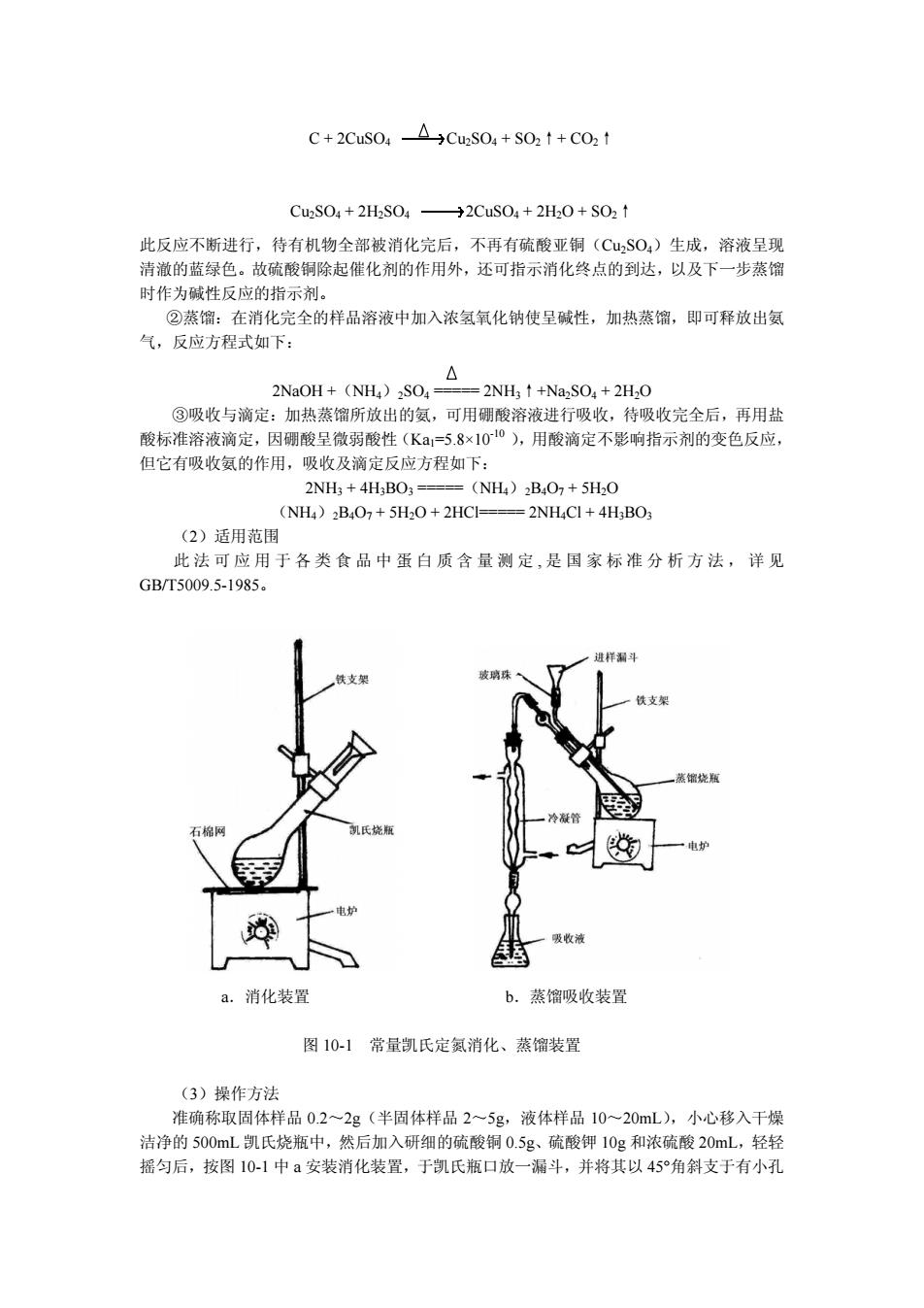
C+2CuSO-ACu:SO+SO:1+CO:t C2S04+2H2S04→2CuS04+2H20+S021 此反应不断进行,待有机物全部被消化完后,不再有硫酸亚铜(CuSO)生成,溶液呈现 清澈的蓝绿色。故硫酸铜除起催化剂的作用外,还可指示消化终点的到达,以及下一步蒸馏 时作为碱性反应的指示剂。 ②燕馏:在消化完全的样品溶液中加入浓氧氧化钠使呈碱性,加热蒸馏,即可释放出氨 气,反应方程式如下: △ 2NaOH+(NH)SO. =2NH2↑+NaSO+2H,0 ③吸收与滴定:加热蒸馏所放出的氨,可用硼酸溶液进行吸收,待吸收完全后,再用盐 酸标准溶液滴定 因硼酸呈微弱酸性(Ka1=5.8×100),用酸滴定不影响指示剂的变色反应 但它有吸收氨的作用,吸收及滴定反应方程如下: 2NH3 +4H3BO3 ==(NH)2B4O7+5H2O (NHA)BO+5HO+2HCI-2NHCI+4HBO (2)话用范围 此法可应用于各类食品中蛋白质含量测定,是国家标准分析方法,详见 GB/T5009.5-1985 a.消化装置 b.蒸馏吸收装置 图10-1常量凯氏定氮消化、蒸馏装置 (3)操作方法 准确称取固体样品0.2~2g(半固体样品2~5g,液体样品10~20mL),小心移入干燥 洁净的500mL凯氏烧瓶中,然后加入研细的硫酸铜0.5g、硫酸钾10g和浓硫酸20mL,轻轻 摇匀后,按图10-1中a安装消化装置,于凯氏瓶口放一漏斗,并将其以45°角斜支于有小孔
C + 2CuSO4 Cu2SO4 + SO2↑+ CO2↑ Cu2SO4 + 2H2SO4 2CuSO4 + 2H2O + SO2↑ 此反应不断进行,待有机物全部被消化完后,不再有硫酸亚铜(Cu2SO4)生成,溶液呈现 清澈的蓝绿色。故硫酸铜除起催化剂的作用外,还可指示消化终点的到达,以及下一步蒸馏 时作为碱性反应的指示剂。 ②蒸馏:在消化完全的样品溶液中加入浓氢氧化钠使呈碱性,加热蒸馏,即可释放出氨 气,反应方程式如下: 2NaOH +(NH4)2SO4 ===== 2NH3↑+Na2SO4 + 2H2O ③吸收与滴定:加热蒸馏所放出的氨,可用硼酸溶液进行吸收,待吸收完全后,再用盐 酸标准溶液滴定,因硼酸呈微弱酸性(Ka1=5.8×10-10 ),用酸滴定不影响指示剂的变色反应, 但它有吸收氨的作用,吸收及滴定反应方程如下: 2NH3 + 4H3BO3 =====(NH4)2B4O7 + 5H2O (NH4)2B4O7 + 5H2O + 2HCl===== 2NH4Cl + 4H3BO3 (2)适用范围 此法可应用于各类食品中蛋白质含量测定 , 是国家标准分析方法,详见 GB/T5009.5-1985。 a.消化装置 b.蒸馏吸收装置 图 10-1 常量凯氏定氮消化、蒸馏装置 (3)操作方法 准确称取固体样品 0.2~2g(半固体样品 2~5g,液体样品 10~20mL),小心移入干燥 洁净的 500mL 凯氏烧瓶中,然后加入研细的硫酸铜 0.5g、硫酸钾 10g 和浓硫酸 20mL,轻轻 摇匀后,按图 10-1 中 a 安装消化装置,于凯氏瓶口放一漏斗,并将其以 45°角斜支于有小孔

的石棉网上。用电炉以小火加热,待内容物全部炭化,泡沫停止产生后,加大火力,保持瓶 内液体微沸,至液体变蓝绿色透明后,再继续加热微沸30min。冷却,小心加入200mL蒸 馏水 ,加入玻璃珠数粒以防蒸馏时暴沸」 将凯氏烧瓶按图10-13 式连好 塞紧瓶口,冷凝管下端插入吸收瓶液面下(瓶 内预先装入50mL40gL硼酸溶液及混合指示剂2~3滴).放松夹子,通过漏斗加入70~80ml 4O0gL氢氧化钠溶液,并摇动凯氏瓶,至瓶内溶液变为深蓝色,或产生黑色沉淀,再加入 100mL蒸馏水(从漏斗中加入),夹紧夹子,加热蒸馏,至氨全部蒸出(馏液约250mL即可), 将冷凝管下端提离液面,用蒸馏水冲洗管口,继续蒸馏lmi,用表面皿接几滴馏出液,以 奈氏试剂检查,如无红棕色物生成, 表示蒸馏完毕, 即可停 上加热 将上述吸收液用0.1000molL盐酸标准溶液直接滴定至由蓝色变为微红色即为终点,记 录盐酸溶液用量,同时作一试剂空白(除不加样品外,从消化开始操作完全相同),记录空 白试验消耗盐酸标准溶液的体积。 (4)结果计算 蛋白质(g100g)= x(-W画 x星x100 式中: -盐酸标准溶液的浓度,moL: 滴定样品吸收液时消耗盐酸标准溶液体积,mL: V E 空白吸收液时消耗盐酸标准溶液体积,mL 样品质量,g M -氨的摩尔质量,14.01gmoL: 下一一氨换算为蛋白质的系数。 (S)说明及注意事项 ①所用试剂溶液应用无氨蒸馏水配制 ②消化时不要用强火,应保持和缓沸腾,以免粘附在凯氏瓶内壁上的含氮化合物在无硫 酸存在的情况下未消化完全而造成氮损失。 ③消化过程中应注意不时转动凯氏烧瓶,以便利用冷凝酸液将附在瓶壁上的固体残渣洗 下并促进其消化完全。 ④样品中若含脂肪或糖较多时,消化过程中易产生大量泡沫,为防止泡沫溢出瓶外,在 开始消化时应用小火加热,并不停地摇动:或者加入少量辛醇或液体石蜡或硅油消泡剂,并 同时注意控制热源强度。 ⑤当样品消化液不易澄清透明时,可将凯氏烧瓶冷却,加入30%过氧化氢2~3mL后再 继续加热消化。 ⑧若取样量较大,如干试样超5,可按每点试样SmL的比例增加硫酸用量 一般消化至呈透明后 ,继续消化30min即可, 但对于含有特别难以氨化的氨化合物 的样品,如含赖氨酸、组氨酸、色氨酸、酪氨酸或脯氨酸等时,需适当延长消化时间。有机 物如分解完全,消化液呈蓝色或浅绿色,但含铁量多时,呈较深绿色。 ⑧蒸馏装置不能漏气。 ©蒸馏前若加诚量不足,消化液呈蓝色不生成氨氧化铜沉淀,此时需再增加氨氧化钠用 最。 ⑩硼酸吸收液的温度不应超过40℃,否则对氨的吸收作用减弱而造成损失,此时可置 于冷水浴中使用。 ①蒸馏完毕后,应先将冷凝管下端提离液面清洗管口,再蒸1mi后关掉热源,否则可
的石棉网上。用电炉以小火加热,待内容物全部炭化,泡沫停止产生后,加大火力,保持瓶 内液体微沸,至液体变蓝绿色透明后,再继续加热微沸 30min。冷却,小心加入 200mL 蒸 馏水,再放冷,加入玻璃珠数粒以防蒸馏时暴沸。 将凯氏烧瓶按图 10-1 蒸馏装置方式连好,塞紧瓶口,冷凝管下端插入吸收瓶液面下(瓶 内预先装入50mL 40g/L硼酸溶液及混合指示剂2~3滴)。放松夹子,通过漏斗加入70~80mL 400g/L 氢氧化钠溶液,并摇动凯氏瓶,至瓶内溶液变为深蓝色,或产生黑色沉淀,再加入 100mL 蒸馏水(从漏斗中加入),夹紧夹子,加热蒸馏,至氨全部蒸出(馏液约 250mL 即可), 将冷凝管下端提离液面,用蒸馏水冲洗管口,继续蒸馏 1min,用表面皿接几滴馏出液,以 奈氏试剂检查,如无红棕色物生成,表示蒸馏完毕,即可停止加热。 将上述吸收液用 0.1000mol/L 盐酸标准溶液直接滴定至由蓝色变为微红色即为终点,记 录盐酸溶液用量,同时作一试剂空白(除不加样品外,从消化开始操作完全相同),记录空 白试验消耗盐酸标准溶液的体积。 (4)结果计算 蛋白质(g/100g)= 式中:c——盐酸标准溶液的浓度,mol/L; V1——滴定样品吸收液时消耗盐酸标准溶液体积,mL; V2——滴定空白吸收液时消耗盐酸标准溶液体积,mL; m——样品质量,g; M 氮——氮的摩尔质量,14.01g/moL; F——氮换算为蛋白质的系数。 (5)说明及注意事项 ①所用试剂溶液应用无氨蒸馏水配制。 ②消化时不要用强火,应保持和缓沸腾,以免粘附在凯氏瓶内壁上的含氮化合物在无硫 酸存在的情况下未消化完全而造成氮损失。 ③消化过程中应注意不时转动凯氏烧瓶,以便利用冷凝酸液将附在瓶壁上的固体残渣洗 下并促进其消化完全。 ④样品中若含脂肪或糖较多时,消化过程中易产生大量泡沫,为防止泡沫溢出瓶外,在 开始消化时应用小火加热,并不停地摇动;或者加入少量辛醇或液体石蜡或硅油消泡剂,并 同时注意控制热源强度。 ⑤当样品消化液不易澄清透明时,可将凯氏烧瓶冷却,加入 30%过氧化氢 2~3mL 后再 继续加热消化。 ⑥若取样量较大,如干试样超过 5g,可按每克试样 5mL 的比例增加硫酸用量。 ⑦一般消化至呈透明后,继续消化 30min 即可,但对于含有特别难以氨化的氮化合物 的样品,如含赖氨酸、组氨酸、色氨酸、酪氨酸或脯氨酸等时,需适当延长消化时间。有机 物如分解完全,消化液呈蓝色或浅绿色,但含铁量多时,呈较深绿色。 ⑧蒸馏装置不能漏气。 ⑨蒸馏前若加碱量不足,消化液呈蓝色不生成氢氧化铜沉淀,此时需再增加氢氧化钠用 量。 ⑩硼酸吸收液的温度不应超过 40℃,否则对氨的吸收作用减弱而造成损失,此时可置 于冷水浴中使用。 ○11 蒸馏完毕后,应先将冷凝管下端提离液面清洗管口,再蒸 1min 后关掉热源,否则可
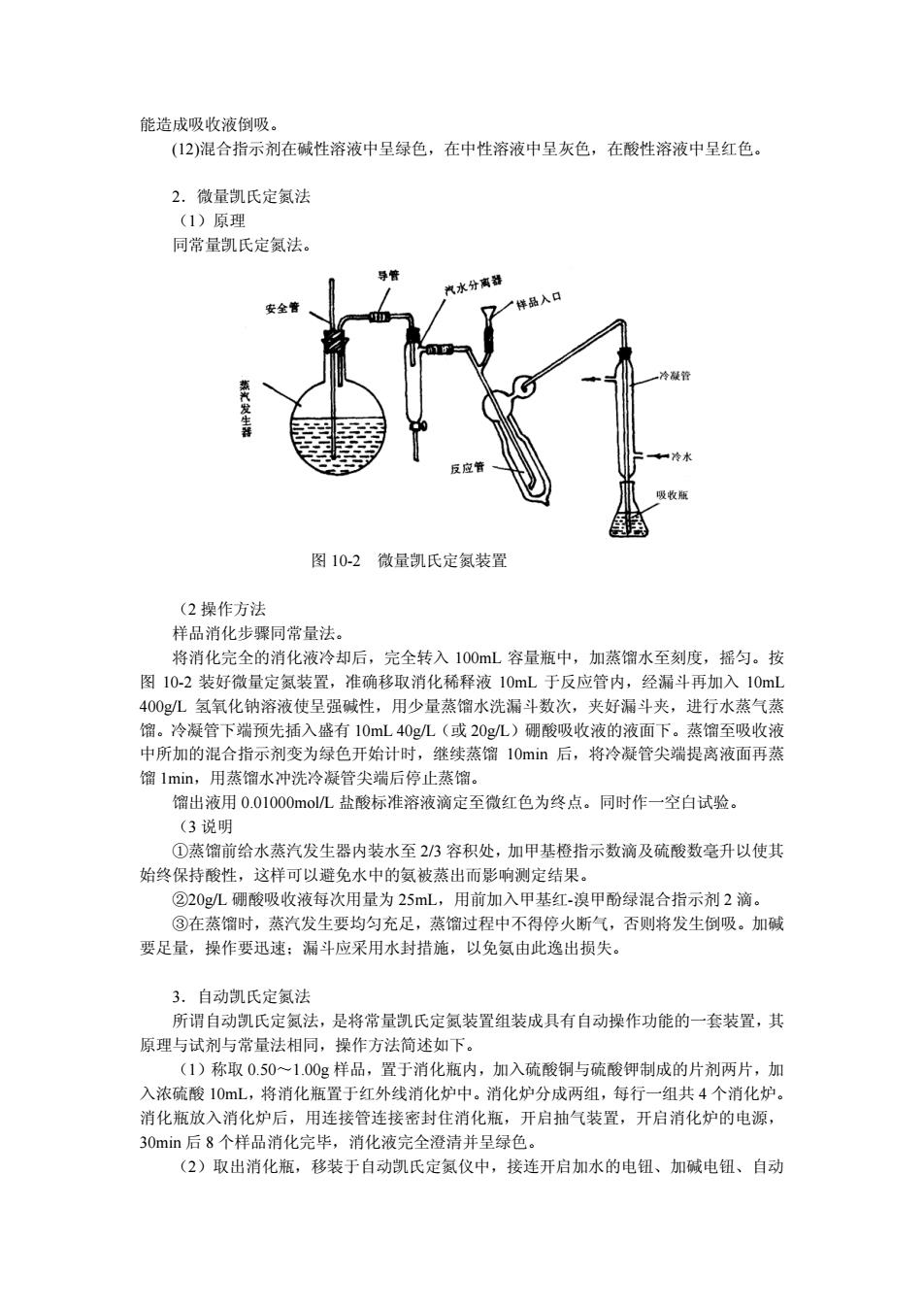
能造成吸收液倒吸 (2)混合指示剂在碱性溶液中呈绿色,在中性溶液中呈灰色,在酸性溶液中呈红色。 2.微量凯氏定氮法 (1)原理 同常量凯氏定氨法。 汽水分离 要全1 图10-2微量凯氏定氨装省 (2操作方法 样品消化步骤同常量法, 将消化完全的消化液冷却后,完全转入100L容量瓶中,加蒸榴水至刻度,摇匀。按 图10,2装好微量定氨装置,准确移取消化稀释液10mL于反应管内,经漏斗再加入10m 400gL氢氧化钠溶液使呈强碱性,用少量蒸馏水洗漏斗数次,夹好漏斗夹,进行水蒸气 馏。冷凝管下端预先插入盛有10mL40eL(或20g/L)硼酸吸收液的液面下。蒸馏至吸收液 中所加的混合指示剂变为绿色开始计时,继续蒸馏10min后,将冷凝管尖端提离液面再蒸 馏lmin,用蒸馏水冲洗冷凝管尖端后停止蒸馏。 榴出液用001000m01盐酸标准溶液滴定至微红伍为终点。同时作一空白试哈 说明 ①蒸馏前给水蒸汽发生器内装水至23容积处,加甲基橙指示数滴及硫酸数毫升以使其 始终保持酸性,这样可以避免水中的氨被蒸出而影响测定结果。 ②20gL硼酸吸收液每次用量为25mL,用前加入甲基红-溴甲酚绿混合指示剂2滴。 ③在蒸馏时,蒸汽发生要均匀充足,蒸馆过程中不得停火断气,否则将发生倒吸。加碱 要足量,操作要迅速:漏斗应采用水封措施,以免氨由此逸出损失 3,自动凯氏定氨法 所谓自动凯氏定氨法,是将常最凯氏定氨装置组装成具有自动操作功能的一套装置,其 原理与试剂与常量法相同,操作方法简述如下。 (1)称取0.50一1.00g样品,置于消化瓶内,加入硫酸铜与硫酸钾制成的片剂两片,加 入浓硫酸10mL,将消化瓶置于红乡 消化 消化炉分成两组 ,每行 一组共 个消化炉 消化瓶放入消化炉后,用连接管连接密封住消化瓶,开启抽气装置,开启消化炉的电源, 30min后8个样品消化完毕,消化液完全澄清并呈绿色。 (2)取出消化瓶,移装于自动凯氏定氨仪中,接连开启加水的电钮、加碱电钮、自动
能造成吸收液倒吸。 (12)混合指示剂在碱性溶液中呈绿色,在中性溶液中呈灰色,在酸性溶液中呈红色。 2.微量凯氏定氮法 (1)原理 同常量凯氏定氮法。 图 10-2 微量凯氏定氮装置 (2 操作方法 样品消化步骤同常量法。 将消化完全的消化液冷却后,完全转入 100mL 容量瓶中,加蒸馏水至刻度,摇匀。按 图 10-2 装好微量定氮装置,准确移取消化稀释液 10mL 于反应管内,经漏斗再加入 10mL 400g/L 氢氧化钠溶液使呈强碱性,用少量蒸馏水洗漏斗数次,夹好漏斗夹,进行水蒸气蒸 馏。冷凝管下端预先插入盛有 10mL 40g/L(或 20g/L)硼酸吸收液的液面下。蒸馏至吸收液 中所加的混合指示剂变为绿色开始计时,继续蒸馏 10min 后,将冷凝管尖端提离液面再蒸 馏 1min,用蒸馏水冲洗冷凝管尖端后停止蒸馏。 馏出液用 0.01000mol/L 盐酸标准溶液滴定至微红色为终点。同时作一空白试验。 (3 说明 ①蒸馏前给水蒸汽发生器内装水至 2/3 容积处,加甲基橙指示数滴及硫酸数毫升以使其 始终保持酸性,这样可以避免水中的氨被蒸出而影响测定结果。 ②20g/L 硼酸吸收液每次用量为 25mL,用前加入甲基红-溴甲酚绿混合指示剂 2 滴。 ③在蒸馏时,蒸汽发生要均匀充足,蒸馏过程中不得停火断气,否则将发生倒吸。加碱 要足量,操作要迅速;漏斗应采用水封措施,以免氨由此逸出损失。 3.自动凯氏定氮法 所谓自动凯氏定氮法,是将常量凯氏定氮装置组装成具有自动操作功能的一套装置,其 原理与试剂与常量法相同,操作方法简述如下。 (1)称取 0.50~1.00g 样品,置于消化瓶内,加入硫酸铜与硫酸钾制成的片剂两片,加 入浓硫酸 10mL,将消化瓶置于红外线消化炉中。消化炉分成两组,每行一组共 4 个消化炉。 消化瓶放入消化炉后,用连接管连接密封住消化瓶,开启抽气装置,开启消化炉的电源, 30min 后 8 个样品消化完毕,消化液完全澄清并呈绿色。 (2)取出消化瓶,移装于自动凯氏定氮仪中,接连开启加水的电钮、加碱电钮、自动

蒸馏滴定电钮,开启电源,大约经12m后由数显装置即可给出样品总氮百分含量,并记 录样品总氨百分比。根据样品的种类选择相应的蛋白质换算系数F,即可得出样品中蛋白质 含量 (3)开启排废液电钮及加水电钮,排出废液并对消化瓶清洗一次 大约在2h左右时间内可完成8个样品的蛋白质含量测定工作。该法具有灵敏、准确、 快速及样品用量少等优点。 二、双缩脲法 1.原理 当脲被小心地加热至150~160℃时,可由两个分子间脱去一个氨分子而生成二缩脲(也 叫双缩眠),反应如下: +H-N(H)-CO-NH2>H2NCONHCONH2+NH, 双缩脲与碱及少量硫酸铜溶液作用生成紫红色的配合物,此反应称为双缩脲反应 OH OH 2 NH-CΓHN NH CuSO 0- C=0 NH 0=C NaoH 0=C C=0 NH: NH-Na NaHaN OH OH (红色配合物 由于蛋白质分子中含有肽键 (-CO-NH-) 与双缩脲结构相似,故也能呈现此反应而生 成紫红色配合物,在一定条件下其颜色深浅与蛋白质含最成正比,据此可用吸收光度法来测 定蛋白质含量,该配合物的最大吸收波长为560nm。 2.方法特点及应用范围 本法灵敏度较低,但操作简单快速,故在生物化学领域中测定蛋白质含量时常用此法 本法亦适用于豆类、油料、米谷等作物种子及肉类等样品测定】 3.操作方法 ①标准曲线的绘制:以采用凯氏定氨法测出蛋白质含量的样品作为标准蛋白质样。按蛋 白质含量40mg、50mg、60mg、70mg、80mg、90mg、100mg、110mg分别称取混合均匀的 标准蛋白质样于8支50mL纳氏比色管中,然后客加入1mL四氯化碳,再用碱性硫酸铜蓉 液准确稀释至50mL,振摇10m ,静置1h,取上层清液离心5min 取离心分离后的透明液 于比色皿中,在560m波长下以蒸馏水作参比液调节仪器零点并测定各溶液的吸光度A 以蛋白质的含量为横坐标,吸光度A为纵坐标绘制标准曲线。 ②样品的测定。准确称取样品适量(即使得蛋白质含量在40~110mg之间)于50mL 纳氏比色管中,加1mL四氯化碳,按上述步骤显色后,在相同条件下测其吸光度A。用测 得的A值在标准曲线上即可查得蛋白质毫克数,进而由此求得样品中的蛋白质含量。 4.结果计算 蛋白质(mg1o0g)=Y1c
蒸馏滴定电钮,开启电源,大约经 12min 后由数显装置即可给出样品总氮百分含量,并记 录样品总氮百分比。根据样品的种类选择相应的蛋白质换算系数 F,即可得出样品中蛋白质 含量。 (3)开启排废液电钮及加水电钮,排出废液并对消化瓶清洗一次。 大约在 2h 左右时间内可完成 8 个样品的蛋白质含量测定工作。该法具有灵敏、准确、 快速及样品用量少等优点。 二、双缩脲法 1.原理 当脲被小心地加热至 150~160℃时,可由两个分子间脱去一个氨分子而生成二缩脲(也 叫双缩脲),反应如下: H2NCONH2 + H-N(H)-CO-NH2 H2NCONHCONH2 + NH3 双缩脲与碱及少量硫酸铜溶液作用生成紫红色的配合物,此反应称为双缩脲反应: O=C NH2 NH O=C NH2 2 CuSO4 NaOH O=C NH2 NH O=C NH2Na OH OH HN C=O C=O NaH2N OH H2N OH Cu (双缩脲) (紫红色配合物) 由于蛋白质分子中含有肽键(-CO-NH-),与双缩脲结构相似,故也能呈现此反应而生 成紫红色配合物,在一定条件下其颜色深浅与蛋白质含量成正比,据此可用吸收光度法来测 定蛋白质含量,该配合物的最大吸收波长为 560nm。 2.方法特点及应用范围 本法灵敏度较低,但操作简单快速,故在生物化学领域中测定蛋白质含量时常用此法。 本法亦适用于豆类、油料、米谷等作物种子及肉类等样品测定。 3.操作方法 ①标准曲线的绘制:以采用凯氏定氮法测出蛋白质含量的样品作为标准蛋白质样。按蛋 白质含量 40mg、50mg、60mg、70mg、80mg、90mg、100mg、110mg 分别称取混合均匀的 标准蛋白质样于 8 支 50mL 纳氏比色管中,然后各加入 1mL 四氯化碳,再用碱性硫酸铜溶 液准确稀释至 50mL,振摇 10min,静置 1h,取上层清液离心 5min,取离心分离后的透明液 于比色皿中,在 560nm 波长下以蒸馏水作参比液调节仪器零点并测定各溶液的吸光度 A, 以蛋白质的含量为横坐标,吸光度 A 为纵坐标绘制标准曲线。 ②样品的测定。准确称取样品适量(即使得蛋白质含量在 40~110mg 之间)于 50mL 纳氏比色管中,加 1mL 四氯化碳,按上述步骤显色后,在相同条件下测其吸光度 A。用测 得的 A 值在标准曲线上即可查得蛋白质毫克数,进而由此求得样品中的蛋白质含量。 4.结果计算 蛋白质(mg/100g)=