
一、 聚合机理 1、概述 1929年,Carothers借用有机化学中加成反应和缩合反应的概念,根据单体和聚合物之 间的组成差异,将聚合反应分为加聚反应和缩聚反应。单体通过相互加成而形成聚合物的反 应称为加聚反应。加聚物具有重复单元和单体分子式结构(原子种类、数目)相同、仅是 子结构(化学键方向、类型)有变化、聚合物相对分子质量是单体相对分子质量整倍数的特 点。带有多个可相互官能团的单体通过有机化学中各种缩合反应消去某些小分子而形成聚合 物的反应称为缩聚反应。 1951年,Foy从聚合反应的机理和动力学角度出发,将聚合反应分为链式聚合和逐步 聚合。链式聚合(也称连锁聚合)需先形成活性中心R,活性中心可以是自由基、阳(正) 离子、阴(负)离子。聚合反应中存在诸如链引发、链增长、链转移、链终止等基元反应,各 基元反应的反应速率和活化能差别很大。链引发是形成活性中心的反应,链增长是大量单体 通过与活性中心的连续加成,最终形成聚合物的过程 单体彼此间不能发生反应,活性中心 失去活性称为链终止。形成一个高分子的反应实际上是在大约一秒钟而且往往是更短的时间 内完成的。反应过程中,反应体系始终由单体、高相对分子质量聚合物和微量引发剂组成, 没有相对分千质量递增的中间物。逐北聚合没有活性中心,它是通过一系列单体上所带的 能相互反应的官能团间的反应逐步实现的。反应中 单体先生成二聚体,再继续反应逐步形 成三聚体、四聚体、五聚体等,直到最后逐步形成聚合物。反应中每一步的反应速率和活化 能大致相同,任何聚合体间均可发生反应。形成一个高分子的反应往往需要数小时。反应过 程中,体系由相对分子质量递的的一系列中间立物所组成。如果进一步别分,结式聚合又可 按活性中心分为自由基聚合、阳离子聚合、阴离子聚合等:而逐步聚合则可按动力学分为平 衡缩聚和不平衡缩聚,如按大分子链结构又可分为线型缩聚和体型缩聚等。 链式聚合与加聚反应,逐步聚合与缩聚反应虽然是从不同的角度进行分类,但两者在许 多情况下经常混用 对于阴子活性聚合来说,其反应有快引发、慢增长、无终止的特点,因此相对分子 量随转化常的提高而线性增大。对于聚氨酯这样单体分子通过反复加成,使分子间形成共价 键,逐步生成高分子聚合物的过程,其反应机理是逐步增长聚合,因此多称为聚加成反应或 逐步加聚反应,但从更)的意义上进,它与生成酚醛树脂的加成缩合反应、生成聚对二甲苯 的氧化偶合反应等都属于逐步聚合。环状单体在聚合反应中环被打开,生成线性聚合物,这 过程称为开环聚合。多数环状单体的开环聚合属于链式聚合。对某些反应,尽管单体和所 得聚合物均相同,但由于反应历程不同,其聚合类型亦不相同。如用己内酰胺合成尼龙6 的反应,用碱为催化剂时属于链式聚合,用酸催化测屈于逐步聚合。因此对聚合反应进行分 类时通常需要兼顾结构和机理 F1oy的分类方法由于涉及到聚合反应本质,得到了人们的青睞。尽管按照聚合反应机 理进行分类有时也有不够明确的地方,但时至今日,对于新的聚合反应,科学家们仍然习惯 于从聚合反应历程进行分类,如活性聚合、开环聚合、异构化聚合、基团转移聚合等。当然 现在的许多新的聚合反应虽然仍可归为某类传统的聚合类型,但其特征已有了明显变化。 2、逐步聚合 逐步聚合的特点是由一系列单体上所带的能相互反应的官能团间的有机反应所组成,在 反应过程中,相互反应的官能团形成小分子而游离于大分子链之外,而单体上相互不反应的

部分测连在一起形成大分子链。利用这一特件,可以很大方便地讲行分子设计,期把日标 物分解为一个个的基本单元,在每个单元上接上可相互反应的活性基团形成单体,再使单体 相互反应即可得到目标产物。 逐步聚合的另一特点是反应的逐步性,一方面由于反应活化能高,体系中一般要加入催 化剂:另一方面由于每一步反成都为平衡反应,因此影响平衡移动的因素都会影响到家北素 合反应 从产物的分子链结构看,缩聚可分为线型缩聚与体型缩聚两大类。 ()线型缩聚 参加聚合反应的单体都只带有两个可相互反应的官能团,聚合讨程中,大分子链成线型 增长,最终得到的聚合物为可溶、培的线型结格 线型缩聚实质上是反应官能团间的反应,从有机化学的角度看,为一系列的平衡反应 对于平衡常数大的线型缩聚,整个聚合在达到所需相对分子质量时反应还未达平衡,这样的 缩聚称为不平衡缩聚:反之称为平衡缩聚 对于不平衡缩聚,产物相对分子质量的控制主要是通过对单体配比的控制来实现。在实 际生产中,往往通过让某一种官能团过量的方法,使最终产物分子链端的官能团失去进一步 反应的能力,以保证在随后的加工、使用过程中聚合物相对分子质量的稳定 对于平衡缩聚,则先要通过排出小分子的办法使平衡往生成聚合物的方向移动,以得到 所需相对分子质量的聚合物 (2)体型缩案 参加聚合的单体中至少有一种含有二个以上可反应的官能团,在反应过程中,分子链 从多个方向进行增长,形成支化和交联的体型聚合物。 为保证聚合反应正常进行,体型缩聚一般分为二或三步进行。第一步聚合形成线型或 支化的相对分子质量较低的预聚物,再进一步反应形成体型聚合物。从预聚物上所带可进 步反应官能团的数目、种类、位置等因素看,如上述因素均比较确定,则称为结构预聚物: 反之则称为无规预聚物。体型缩聚的一个关健是轻制反应停止于预聚物,以防止凝胶的生成 3、连锁聚合 连锁聚合的一个重要特点是存在活性中心R”,它一般是通过加入引发剂(或催化剂) 产生的。依活性中心的不同,连锁聚合可以进一步划分为自由基聚合、阳(正)离子聚合、阴 负)离子聚合 ()自由基聚合 活性中心是自由基的连锁聚合称自由基聚合机理。自由基聚合的特征是慢引发、快增长、 右转移、速终止。 自由堪聚合可采用引发剂回引发、热引发、光引发、辐射引发等。实际中多采用引发剂引 发,常用的有偶氨类引发剂、过氧类引发剂、氧化还原类引发剂等。引发剂的选择十分关 键,往往决定一个聚合反应的成败。 链增长阶段多存在自动加速现象,这是由于随转化率提高,体系粘度加人,阻止了链 自由甚云动,使双基斜终止反应几离下降成的 自由基聚合中存在大量的链转移反应, 主要为向单体、溶剂、引发剂、聚合物的转移 反应。链转移可造成相对分子质量下降,产生支链、引发效率下降等。利用这一特性,可加 入链转移常数适当的物质作为链转移剂调节聚合物相对分子质量。 正常情况下自由基聚合为双基终止,有偶合终止和歧化终止两种 (2)阴离子聚合
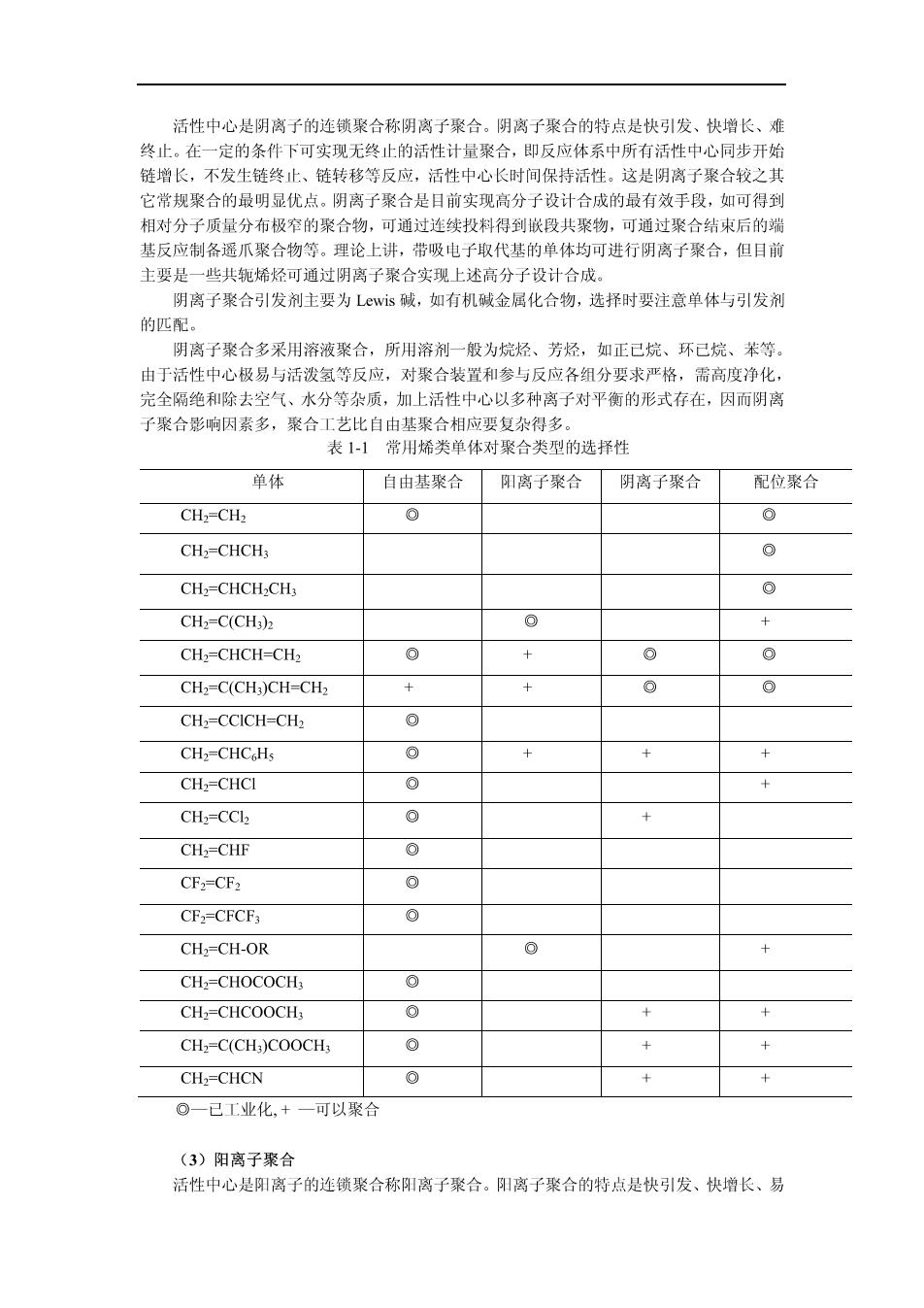
活性中心是阴离子的连锁聚合称阴离子聚合。阴离子聚合的特点是快引发、快增长、难 终止。在一定的条件下可实现无终止的活性计量聚合,即反应体系中所有活性中心同步开始 链增长,不发生链终止、链转移等反应,活性中心长时间保持活性。这是阴离子聚合较之其 它常规聚合的最明显优点。阴离子聚合是目前实现高分子设计合成的最有效手段,如可得到 相对分子质量分布极窄的聚合物,可通过连续投料得到嵌段共聚物,可通讨聚合结束后的瑞 基反应制各遥爪聚合物等。理论上讲,带吸电子取代基的单体均可进行阴离子聚合,但目前 主要是一些共轭烯烃可通过阴离子聚合实现上述高分子设计合成。 阴离子聚合引发剂主要为Lwis碱,如有机碱金属化合物,选择时要注意单体与引发剂 的匹配 阴离子聚合多采用溶液聚合,所用溶剂一般为烷烃、芳烃,如正己烷、环己烷、苯等。 由于活性中心极易与活泼氢等反应,对聚合装置和参与反应各组分要求严格,需高度净化 完全隔绝和除去空气、水分等杂质,加上活性中心以多种离子对平衡的形式存在,因而阴离 子聚合影响因素多,聚合工艺比自由基聚合相应要复杂得多。 表1-1常用烯类单体对聚合类型的选择性 单体 自由基聚合 阳离子聚合 阴离子聚合 配位聚合 CH2-CH: CH,-CHCH CH=CHCH-CH @ CH2=C(CH) e CH,=CHCH-CH> 0 0 CH2=C(CH )CH=CH> 十 CH2=CCICH=CH2 0 CH2=CHCoHs 0 CH2=CHCI CH2=CCl 0 CH2=CHF 0 CF2=CF2 CF2-CFCF3 CHz=CH-OR CH-CHOCOCH CH=CHCOOCH 0 CHz-C(CH)COOCH 0 CH2=CHCN 0 @一已工业化.+一可以聚合 (3)阳离子聚合 活性中心是阳离子的连锁聚合称阳离子聚合。阳离子聚合的特点是快引发、快增长、易

转移、难终止,由于反应活化能低,链转移严重,为此阳离子聚合多采用低温聚合(如聚异 烯需在-100℃进行聚合 ,以得高相对分子质量聚合物,所以除少数只能进行阳离子聚合 的单体,如异丁烯、烷基乙烯基醚等,一般不采用阳离子聚合。 阳离子聚合引发剂主要有L©is酸(多用于高相对分子质量聚合物合成)和质子酸。 阳离子聚合多采用溶液聚合,溶剂一般为极性溶剂,如卤代烷烃。其它方面与阴离子聚 合类似,聚合工艺控制复杂 (4)配位聚合 配位聚合的概念最初是Nata在解释a-烯烃用Ziegler-Nata催化剂聚合的聚合机理时提 的。是指单体分子的碳碳双键先在讨渡金屈雅化剂的活件中心的必位上配位,形成某种 形式的络 (常称 严络合物),随后单体分子相继插入过渡金属-碳键中进行增长,因 此又称络合聚合。配位聚合最重要的是其催化体,一般称Ziegler-Natta催化剂,主要特点是 可合成出立构规整聚合物。 在实际应用中,配位与离子型聚合有许多相似之处,如要求体系密闭,去除空气和水! 原科需要精制,反应需在氨气保护下进行等。这是由于Ziegler-Natta催化剂易与空气和水发 生副反应而失活, (5)开环聚合 环状单体在聚合过程中通过不断地开环反成形成高聚物的过程称开环聚合, 能够进行开环聚合的单体很多,如环状烯烃,以及内酯、内酰胺、环醚、环硅氧烷等环 内含 个或多个杂原子的杂环化合物。环状单体能否转变为聚合物,从热力学角度分析 取决于聚合过程中自由能的变化情况,与环状单体和线形聚合物的相对稳定性有关。一般而 言,六元环相对稳定不能聚合,其它环烷经的聚合可行性为:三元环,四元环>八元环>五 元环,七元环。对于三元环、四元环来讲,△H是决定△G的主要因素:而对于五元环 六元环和七元环米说,△H°和△S°对AG°的贡献都重要。随着环节数的增加,嫡变对自 由能变化的贡献增大,十二元环以上的环状单体,熵变是开环聚合的主要推动力。对于环烷 烃来讲,取代基的存在将降低聚合反应的热力学可行性。在线形聚合物中,取代基的相互作 用要比在环状单体中的大,△H变大(向正值方向变化),△S变小,使得聚合倾向变小 从动力学角度分析,在环烷烃的结构中由于不存在容易被引发物种进攻的键 因此开环聚台 难丁进行。内酰胺、内酯、环醚及其他的环状单体由于杂原子的存在提供了可接受引发物 亲核或亲电进攻的部位,从而可以进行开环聚合的引发及增长反应。总的说来,三元、四元 和七到十一元环的可聚性高,而五、六元环的可聚性低。实际上开环聚合一般仅限于九元环 以下的环状单体, 更人的环状单体一般是不容易得到的 开环聚合既具有某些加成聚合的特征,也具有缩合聚合的特征。开环聚合从表面上看, 也存在若链引发、链增长、链终止等基元反应:在增长阶段,单体只与增长链反应。这一点 与连锁聚合相似。但开环聚合也具有逐步聚合的特征,即在聚合过程中,聚合物的平均分子 质量随聚合的进行而增长,区分逐步 聚合和连锁聚合的主要标志是聚合物的平均分 子质量健 聚合时间的变化情况。逐步聚合中,平均分子质量随聚合反应的进行增长缓慢:而连锁聚合 的整个过程中都有高聚物生成,聚合体系中只存在高聚物、单体及少量的增长链,单体只能 增长链反应。大数的开环聚合为深步聚合,也有些是完全的作镜聚合。开环聚合大名为 离子型聚合,如增长链存在着离子对,反应速度受溶剂的影响等。许多开环聚合还具有活性 聚合的特征。 开环聚合与缩聚反应相比,还具有紧合条件温和、能够自动保持官能团等物质的量等特 点,因此开环聚合所得聚合物的平均分子质量,通常要比缩聚物高得多:另一方面,开环聚 合可供选择的单体比缩聚反应少,加上有些环状单体合成困难,因此由开环聚合所得到的聚
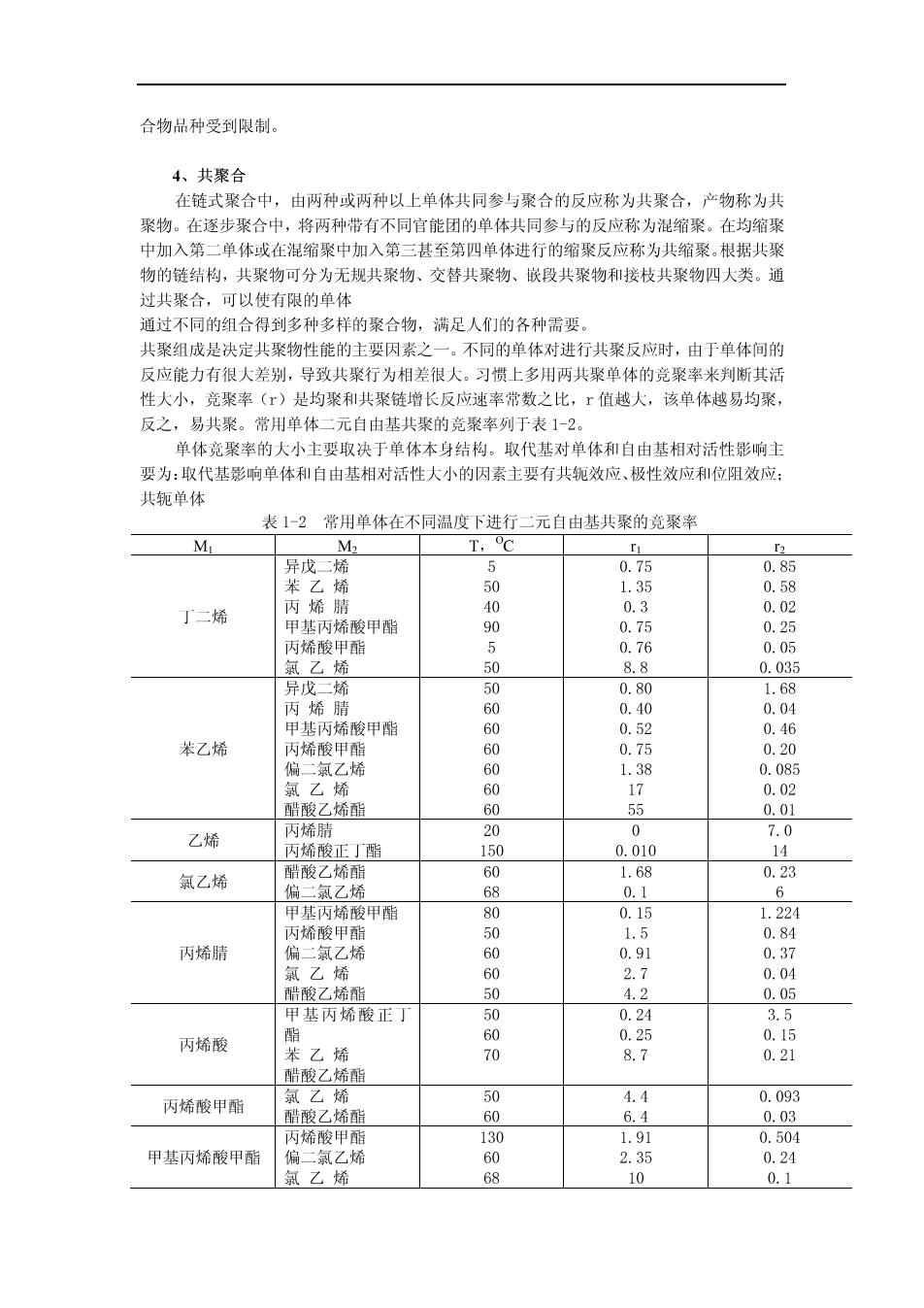
合物品种受到限制。 4、共聚合 在链式聚合中,由两种或两种以上单体共同参与聚合的反应称为共聚合,产物称为共 聚物。在逐步聚合中,将两种带有不同官能团的单体共同参与的反应称为混缩聚。在均缩聚 中加入第二单体或在混缩聚中加入第三甚至第四单体进行的缩聚反应称为共缩聚。根据共聚 物的链结构,共聚物可分为无规共聚物、交替共聚物、嵌段共聚物和接枝共聚物四大类。通 过共聚合,可以使有限的单体 通过不同的组合得到多种多样的聚合物,满足人们的各种需要。 共聚组成是决定共聚物性能的主要因素之一,不同的单体对进行共聚反应时,由于单体间的 反应能力有很大差别,导致共聚行为相差很大。习惯上多用两共聚单体的竞聚率米判断其活 性人小,竞聚率()是均聚和共聚链增长反应速率常数之比,「值越人,该单体越易均聚 反之,易共聚。常用单体二元自由基共聚的竞聚率列于表12。 单体音聚率的大小主要取决干单体本身结构。取代其时单体和白由其相对活性影临主 要为:取代基影响单体和自由基相对活性大小的因素主要有共轭效应、极性效应和位阻效应 共轭单体 表1-2常用单体在不同温度下进行二元自由基共聚的竞聚率 T.C 异戊二烯 0.75 0.85 丁二烯 甲基丙烯酸甲酯 丙稀酸甲酯 55040005 氯乙 88 0.03s 异戊二烯 基丙烯酸甲醒 苯乙烯 偏 乙烯 6 0.02 酷酸乙烯酯 0.0 乙烯 正丁 氯乙烯 08 0.1 甲基丙烯酸甲酯 0.1 1.22 丙烯酸甲酯 0.84 丙烯睛 乙烯 甲基丙烯酸正了 0 2 丙烯酸 0. 0.15 苯乙烯 8.7 0.21 丙烯酸甲酯 50 0504 甲基丙烯酸甲酯 0.24 氯乙烯 68 10 0.1